Kun hyönteisgenomia selvitettiin, havaittiin, että. Genomi genomissa: vanha loinen. Vaikeudet kasvien genomien tutkimisessa
DNA:n rakenteen löytämisen 50-vuotispäivää
A.V. Zelenin
KASVIGENOME
A. V. Zelenin
Zelenin Aleksanteri Vladimirovitš- d.b.n.,
molekyylibiologian instituutin laboratorion johtaja. V.A. Engelhardt RAS.
"Human Genome" -ohjelman vaikuttavat saavutukset sekä ns. ekstrapienten (virukset), pienten (bakteerit, hiiva) ja keskikokoiset (suolimato, Drosophila) genomien salaustyön onnistuminen mahdollistivat siirtyä laajamittaiseen suurten ja erittäin suurien kasvien genomien tutkimukseen. Taloudellisesti tärkeimpien kasvien genomien yksityiskohtaisen tutkimuksen kiireellistä tarvetta korostettiin Yhdysvalloissa vuonna 1997 pidetyssä kasvigenomiikkaa käsittelevässä kokouksessa [ , ]. Siitä kuluneiden vuosien aikana tällä alalla on saavutettu kiistattomia menestyksiä. Vuonna 2000 ilmestyi julkaisu pienen sinapin - Arabidopsiksen - genomin täydellisestä sekvensoinnista (koko ydin-DNA:n lineaarisen nukleotidisekvenssin määritys), vuonna 2001 - riisin genomin alustavasta (luonnos) sekvensoinnista. Suurten ja supersuurien kasvigenomien (maissi, ruis, vehnä) sekvensointitöitä raportoitiin toistuvasti, mutta nämä raportit eivät sisältäneet erityistä tietoa ja olivat pikemminkin aiejulistuksia.
Kasvien genomien dekoodauksen oletetaan avaavan laajat mahdollisuudet tieteelle ja käytännölle. Ensinnäkin uusien geenien tunnistaminen ja niiden geneettisen säätelyn ketju lisäävät merkittävästi kasvien tuottavuutta bioteknisten lähestymistapojen avulla. Geenien löytäminen, eristäminen, lisääntyminen (kloonaus) ja sekvensointi, jotka vastaavat kasviorganismin sellaisista tärkeistä toiminnoista kuin lisääntyminen ja tuottavuus, vaihteluprosessit, vastustuskyky haitallisille ympäristötekijöille sekä kromosomien homologinen pariutuminen, syntyi liittyy uusia mahdollisuuksia jalostusprosessin parantamiseen. Lopuksi eristettyjä ja kloonattuja geenejä voidaan käyttää siirtogeenisten kasvien saamiseksi, joilla on täysin uusia ominaisuuksia, ja analysoida geeniaktiivisuuden säätelymekanismeja.
Kasvigenomien tutkimisen tärkeyttä korostaa myös se, että toistaiseksi lokalisoitujen, kloonattujen ja sekvensoitujen kasvigeenien määrä on pieni ja vaihtelee eri arvioiden mukaan 800 ja 1200 välillä. Tämä on 10-15 kertaa vähemmän kuin vuonna 2010. esimerkiksi ihmisissä.
Yhdysvallat on edelleen kiistaton johtaja kasvien genomien laajamittaisessa tutkimuksessa, vaikka riisin genomin intensiivisiä tutkimuksia tehdään Japanissa ja viime vuodet ja Kiinassa. Arabidopsiksen genomin tulkitsemiseen osallistuivat yhdysvaltalaisten laboratorioiden lisäksi aktiivisesti eurooppalaiset tutkimusryhmät. Yhdysvaltojen näennäinen johtajuus aiheuttaa vakavaa huolta eurooppalaisissa tutkijoissa, minkä he ilmaisivat selvästi kokouksessa merkittävällä otsikolla "Prospects for Genomics in the Post-Genomic Era", joka pidettiin vuoden 2000 lopulla Ranskassa. Amerikkalaisen tieteen edistyminen maatalouskasvien genomien tutkimisessa ja siirtogeenisten kasvimuotojen luomisessa eurooppalaisten tutkijoiden mukaan uhkaa, että ei liian kaukaisessa tulevaisuudessa (kahdesta viiteen vuosikymmeneen), jolloin väestönkasvu asettaa ihmiskunnan yleisen tilanteen eteen. elintarvikekriisi, Euroopan talous ja tiede tulevat riippuvaisiksi amerikkalaisesta teknologiasta. Tältä osin ilmoitettiin ranskalais-saksalaisen tieteellisen ohjelman perustamisesta kasvien genomien tutkimukseen ("Plantgene") ja siihen tehtiin merkittäviä investointeja.
Ilmeisesti kasvien genomiikan ongelmien tulisi kiinnittää venäläisten tutkijoiden ja tieteen järjestäjien sekä hallintoelinten tarkkaavainen huomio, koska kyse ei ole vain tieteellisestä arvovallasta, vaan myös maan kansallisesta turvallisuudesta. Vuosikymmenen tai kahden kuluttua ruoasta tulee tärkein strateginen resurssi.
KASVIGENOMIEN TUTKIMUKSEN VAIKKEUDET
Kasvien genomien tutkiminen on paljon vaikeampi tehtävä kuin ihmisten ja muiden eläinten genomin tutkiminen. Tämä johtuu seuraavista olosuhteista:
valtavat genomikoot, jotka saavuttavat kymmeniä ja jopa satoja miljardeja emäspareja (bp) yksittäisten kasvilajien kohdalla: tärkeimpien taloudellisesti tärkeiden kasvien (riisi, pellava ja puuvilla) genomit ovat joko kooltaan lähellä ihmisen genomia tai ylittää sen monta kertaa (taulukko);GENOMIEN KROMOSOMITUTKIMUKSETKromosomien lukumäärän voimakkaat vaihtelut eri kasveissa - kahdesta joissakin lajeissa useisiin satoihin toisissa, eikä genomin koon ja kromosomien lukumäärän välillä ole mahdollista tunnistaa tiukkaa korrelaatiota;
Lukuisat polyploidimuodot (jotka sisältävät enemmän kuin kaksi genomia solua kohden) muodostavat samanlaisia, mutta eivät identtisiä genomeja (alpolyploidia);
Kasvien genomien äärimmäinen rikastuminen (jopa 99 %) "merkittämättömästä" (ei-koodaavasta, eli geenejä sisältämättömästä) DNA:sta, mikä tekee sekvensoitujen fragmenttien erittäin vaikeaksi liittyä (järjestyä oikeaan järjestykseen) yhteiseksi suurikokoinen DNA-alue (jatkuvuus);
Epätäydellinen (verrattuna Drosophilan, ihmisen ja hiiren genomiin) morfologinen, geneettinen ja fyysinen kromosomien kartoitus;
Käytännön mahdottomuus eristää yksittäisiä kromosomeja puhtaassa muodossa menetelmillä, joita tavallisesti käytetään tähän tarkoitukseen ihmisten ja eläinten kromosomeille (lajittelu virrassa ja soluhybridien käyttö);
Yksittäisten geenien kromosomikartoituksen (paikan määrittäminen kromosomissa) vaikeus hybridisaatiolla paikan päällä johtuen sekä "merkittämättömän" DNA:n suuresta pitoisuudesta kasvin genomissa ja sen erityispiirteistä rakenteellinen organisaatio kasvien kromosomit;
Kasvien evoluutionaalinen etäisyys eläimistä, mikä vaikeuttaa vakavasti ihmisten ja muiden eläinten genomien sekvensoimalla saadun tiedon käyttöä kasvien genomien tutkimiseen;
Useimpien kasvien pitkä lisääntymisprosessi, mikä hidastaa merkittävästi niiden geneettistä analyysiä.
Yleensä genomien ja erityisesti kasvien kromosomaalisilla (sytogeneettisillä) tutkimuksilla on pitkä historia. Termillä "genomi" ehdotettiin haploidista (yksittäistä) kromosomien sarjaa niiden sisältämien geenien kanssa 1900-luvun ensimmäisellä neljänneksellä, toisin sanoen kauan ennen kuin DNA:n rooli geneettisen tiedon kantajana vakiintui. .
Uuden, aiemmin geneettisesti tutkimattoman monisoluisen organismin genomin kuvaus alkaa yleensä sen kromosomien täydellisen sarjan (karyotyypin) tutkimisesta ja kuvauksesta. Tämä koskee tietysti myös kasveja, joista suurta määrää ei ole edes aloitettu tutkimaan.
Jo kromosomitutkimusten kynnyksellä verrattiin sukulaiskasvilajien genomeja interspesifisten hybridien meioottisen konjugaation (homologisten kromosomien yhdistelmän) analyysin perusteella. Viimeisten 100 vuoden aikana kromosomianalyysin mahdollisuudet ovat laajentuneet dramaattisesti. Nyt kasvien genomien karakterisointiin käytetään kehittyneempiä tekniikoita: niin sanotun differentiaalivärjäyksen erilaisia variantteja, jotka mahdollistavat morfologiset ominaisuudet tunnistaa yksittäiset kromosomit; hybridisaatio paikan päällä spesifisten geenien paikallistamisen mahdollistaminen kromosomeihin; soluproteiinien biokemialliset tutkimukset (elektroforeesi ja immunokemia) ja lopuksi joukko menetelmiä, jotka perustuvat kromosomaalisen DNA:n analyysiin sen sekvensointiin asti.

Riisi. 1. Viljan karyotyypit a - ruis (14 kromosomia), b - durumvehnä (28 kromosomia), c - pehmeä vehnä (42 kromosomia), d - ohra (14 kromosomia)Viljojen, pääasiassa vehnän ja rukiin, karyotyyppejä on tutkittu useiden vuosien ajan. Mielenkiintoista on, että näiden kasvien eri lajeissa kromosomien lukumäärä on erilainen, mutta aina seitsemän kerrannainen. Yksittäiset viljatyypit voidaan tunnistaa luotettavasti niiden karyotyypin perusteella. Esimerkiksi rukiin genomi koostuu seitsemästä parista suuria kromosomeja, joiden päissä on voimakkaan värisiä heterokromaattisia lohkoja, joita usein kutsutaan segmenteiksi tai vyöhykkeiksi (kuva 1a). Vehnägenomeissa on jo 14 ja 21 kromosomiparia (kuvat 1, b, c), ja heterokromaattisten lohkojen jakautuminen niissä ei ole sama kuin rukiin kromosomeissa. Yksittäiset vehnän genomit, merkinnällä A, B ja D, eroavat myös toisistaan. Kromosomien lukumäärän kasvu 14:stä 21:een johtaa vehnän ominaisuuksien jyrkkään muutokseen, mikä näkyy niiden nimissä: durum, tai pastaa, vehnää ja pehmeää, tai leipää, vehnää . Gluteeniproteiinien geenejä sisältävä D-geeni, joka antaa taikinalle ns. itävyyden, vastaa siitä, että pehmeä vehnä saa korkeat leivontaominaisuudet. Juuri tähän genomiin kiinnitetään erityistä huomiota leipävehnän valinnan parantamisessa. Toista 14-kromosomista viljaa, ohraa (kuva 1, d), ei yleensä käytetä leivän valmistukseen, mutta se on pääraaka-aine yleisten tuotteiden kuten oluen ja viskin valmistuksessa.
Joidenkin tärkeimpien maatalouslajien, kuten vehnän luonnonvaraisten sukulaisten - Aegilopsin, laadun parantamiseen käytettyjen luonnonvaraisten kasvien kromosomeja tutkitaan intensiivisesti. Uusia kasvimuotoja luodaan risteyttämällä (kuva 2) ja valinnalla. Viime vuosina tutkimusmenetelmien merkittävä parannus on mahdollistanut sellaisten kasvigenomien tutkimuksen aloittamisen, joiden karyotyyppien ominaisuudet (lähinnä kromosomien pieni koko) tekivät niistä aiemmin saavuttamattomissa kromosomianalyysissä. Joten vasta äskettäin kaikki puuvillan, kamomillan ja pellavan kromosomit tunnistettiin ensimmäistä kertaa.

Riisi. 2. Vehnän karyotyypit ja vehnän hybridi Aegilopsin kanssa
a - heksaploidinen pehmeä vehnä ( Triticum astivum), joka koostuu A-, B- ja O-genomeista; b - tetraploidinen vehnä ( Triticum timopheevi), joka koostuu A- ja G-genomeista. sisältää geenejä, jotka ovat vastustuskykyisiä useimmille vehnäsairauksille; c - hybridit Triticum astivum X Triticum timopheevi vastustuskykyinen härmäsientä ja ruostetta vastaan, osan kromosomista uusiutuminen on selvästi näkyvissäDNA:N ENSISIJAINEN RAKENNE
Molekyyligenetiikan kehittymisen myötä genomin käsite on laajentunut. Nyt tätä termiä tulkitaan sekä klassisessa kromosomaalisessa että nykyaikaisessa molekyylisessä mielessä: yksittäisen viruksen, solun ja organismin koko geneettinen materiaali. Luonnollisesti tutkimuksen jälkeen täydellinen ensisijainen rakenne genomit (tätä kutsutaan usein täydelliseksi lineaariseksi emästen sekvenssiksi nukleiinihapot) useiden mikro-organismien ja ihmisten osalta nousi esiin kysymys kasvien genomien sekvensoinnista.
Monista kasviorganismeista kaksi valittiin tutkimukseen - Arabidopsis, joka edustaa kaksisirkkaisten luokkaa (genomin koko 125 miljoonaa bp), ja riisi yksisirkkaisten luokasta (420-470 miljoonaa bp). Nämä genomit ovat pieniä verrattuna muihin kasvigenomeihin ja sisältävät suhteellisen vähän toistuvia DNA-segmenttejä. Tällaiset ominaisuudet antoivat toivoa, että valitut genomit olisivat käytettävissä niiden primäärirakenteen suhteellisen nopeaa määritystä varten.

Riisi. 3. Arabidopsis - pieni sinappi - pieni kasvi ristikukkaisten heimosta ( Brassicaceae). Lehden yhden sivun pinta-alalla voit kasvattaa jopa tuhatta yksittäistä Arabidopsis-organismia.Syynä Arabidopsiksen valintaan ei ollut vain sen genomin pieni koko, vaan myös organismin pieni koko, mikä tekee siitä helppoa kasvattaa laboratoriossa (kuva 3). Otimme huomioon sen lyhyen lisääntymissyklin, jonka ansiosta on mahdollista tehdä nopeasti risteyttämis- ja valintakokeita, yksityiskohtaisesti tutkittu genetiikka, käsittelyn helppous muuttuvissa kasvuolosuhteissa (muutokset maaperän suolakoostumuksessa, erilaisten lisäys). ravinteita jne.) ja erilaisten mutageenisten tekijöiden ja taudinaiheuttajien (virukset, bakteerit, sienet) vaikutusten testaaminen kasveihin. Arabidopsiksella ei ole taloudellista arvoa, joten sen genomia kutsuttiin hiiren genomin ohella viitteeksi tai, vähemmän tarkasti, malliksi.*
* Termin "malligenomi" esiintyminen venäläisessä kirjallisuudessa on seurausta englanninkielisen lauseen malli genomi virheellisestä käännöksestä. Sana "malli" ei tarkoita vain adjektiivia "malli", vaan myös substantiivia "näyte", "standardi", "malli". Olisi oikeampaa puhua näytegenomista tai referenssigenomista.Intensiivinen työ Arabidopsis-genomin sekvensointiin aloitti vuonna 1996 kansainvälinen konsortio, johon kuului tieteellisiä instituutioita ja tutkimusryhmiä Yhdysvalloista, Japanista, Belgiasta, Italiasta, Isosta-Britanniasta ja Saksasta. Joulukuussa 2000 saatiin laajaa tietoa Arabidopsis-genomin primäärirakenteen määrittämisestä. Sekvensoinnissa käytettiin klassista tai hierarkkista tekniikkaa: ensin tutkittiin yksittäisiä pieniä genomin osia, joista koostui suurempia osia (contigeja) ja viimeisessä vaiheessa yksittäisten kromosomien rakennetta. Arabidopsis-genomin ydin-DNA on jakautunut viiteen kromosomiin. Vuonna 1999 julkaistiin kahden kromosomin sekvensoinnin tulokset, ja lehdistössä ilmestynyt tieto kolmen jäljellä olevan kromosomin primäärirakenteesta saattoi koko genomin sekvensoinnin päätökseen.
125 miljoonasta emäsparista on määritetty 119 miljoonan primäärirakenne, mikä on 92 % koko genomista. Vain 8 % Arabidopsis-genomista, joka sisälsi suuria lohkoja toistuvia DNA-segmenttejä, osoittautui mahdottomaksi tutkia. Eukaryoottisen genomin sekvensoinnin täydellisyyden ja perusteellisuuden suhteen Arabidopsis on edelleen kolmen parhaan mestarin joukossa yksisoluisen hiivaorganismin kanssa. Saccharomyces cerevisiae Ja monisoluinen organismi eläin Caenorhabditis eleganssia(katso taulukko).
Arabidopsiksen genomista on löydetty noin 15 000 yksittäistä proteiinia koodaavaa geeniä. Näistä noin 12 000 on kahtena kopiona haploidista (yksittäistä) genomia kohden, joten kokonaismäärä geenejä on 27 000. Arabidopsiksen geenien määrä ei juuri eroa organismien, kuten ihmisen ja hiiren, geenien lukumäärästä, mutta sen genomin koko on 25-30 kertaa pienempi. Tämä seikka liittyy tärkeisiin piirteisiin Arabidopsiksen ja yksittäisten geenien rakenteessa yleinen rakenne hänen genominsa.
Arabidopsis-geenit ovat kompakteja, ja ne sisältävät vain muutaman eksonin (proteiinia koodaavia alueita), jotka on erotettu lyhyillä (noin 250 bp) ei-koodaavilla DNA-segmenteillä (introneilla). Yksittäisten geenien väliset välit ovat keskimäärin 4600 emäsparia. Vertailun vuoksi huomautamme, että ihmisen geenit sisältävät useita kymmeniä ja jopa satoja eksoneja ja introneita, ja geenien välisten alueiden koko on 10 tuhatta emäsparia tai enemmän. Oletetaan, että pienen kompaktin genomin läsnäolo vaikutti Arabidopsiksen evolutionaariseen stabiilisuuteen, koska sen DNA:sta tuli vähemmässä määrin kohde useille vahingollisille aineille, erityisesti viruksen kaltaisten toistuvien DNA-fragmenttien (transposonien) viemiselle. genomiin.
Muiden Arabidopsis-genomin molekyyliominaisuuksien joukossa on huomattava, että eksonit ovat rikastettuja guaniinilla ja sytosiinilla (44 % eksoneissa ja 32 % introneissa) verrattuna eläingeeneihin, sekä kaksinkertaisesti toistuvien (kaksoistettujen) geenien läsnäolo. Oletetaan, että tällainen kaksinkertaistuminen tapahtui neljän samanaikaisen tapahtuman seurauksena, jotka koostuvat Arabidopsis-geenien osan kaksinkertaistumisesta (toistosta) tai sukulaisten genomien fuusiosta. Nämä tapahtumat, jotka tapahtuivat 100-200 miljoonaa vuotta sitten, ovat ilmentymä yleisestä polyploidisaatiotrendistä (organismin genomien lukumäärän moninkertainen lisääntyminen), joka on tyypillistä kasvigenomeille. Jotkut tosiasiat osoittavat kuitenkin, että Arabidopsiksen kaksoisgeenit eivät ole identtisiä ja toimivat eri tavalla, mikä voi liittyä mutaatioihin niiden säätelyalueilla.
Riisistä on tullut toinen täydellisen DNA-sekvensoinnin kohde. Tämän kasvin genomi on myös pieni (12 kromosomia, yhteensä 420-470 miljoonaa bp), vain 3,5 kertaa suurempi kuin Arabidopsiksen. Toisin kuin Arabidopsis, riisillä on kuitenkin suuri taloudellinen merkitys, sillä se on ravinnon perusta yli puolelle ihmiskunnasta, joten miljardien kuluttajien lisäksi myös monimiljoonainen armeija osallistuu aktiivisesti sen erittäin työläiseen prosessiin. viljely on elintärkeästi kiinnostunut sen ominaisuuksien parantamisesta.
Jotkut tutkijat alkoivat tutkia riisin genomia jo 1980-luvulla, mutta nämä tutkimukset saavuttivat vakavan mittakaavan vasta 1990-luvulla. Vuonna 1991 Japanissa luotiin ohjelma riisin genomin rakenteen tulkitsemiseksi, ja se kokosi yhteen monien tutkimusryhmien ponnistelut. Tämän ohjelman pohjalta järjestettiin vuonna 1997 International Rice Genome Project. Sen osallistujat päättivät keskittää ponnistelunsa riisin yhden alalajin sekvensointiin ( Oriza sativajaponica), jonka tutkimuksessa oli jo tuolloin saavutettu merkittävää edistystä. Vakava ärsyke ja kuvaannollisesti sanottuna johtotähti sellaiselle työlle oli "Human Genome" -ohjelma.
Tämän ohjelman puitteissa testattiin genomin "kromosomaalisen" hierarkkisen jaon strategiaa, jota kansainvälisen konsortion osallistujat käyttivät riisin genomin tulkinnassa. Kuitenkin, jos ihmisen genomin tutkimuksessa eristettiin yksittäisten kromosomien fraktioita eri menetelmillä, niin yksittäisille riisikromosomeille ja niiden yksittäisille alueille spesifinen materiaali saatiin lasermikrodissektiolla (mikroskooppisten esineiden leikkaaminen). Mikroskoopin objektilasilla, jossa on riisin kromosomit sijaitsevat, lasersäteen vaikutuksesta kaikki palaa, paitsi kromosomi tai sen analyysiin varatut osat. Jäljelle jäävää materiaalia käytetään kloonaukseen ja sekvensointiin.
Lukuisia raportteja on julkaistu riisin genomin yksittäisten fragmenttien sekvensoinnin tuloksista, jotka on suoritettu suurella tarkkuudella ja yksityiskohtaisesti, mikä on ominaista hierarkkiselle teknologialle. Riisigenomin täydellisen primäärirakenteen uskottiin valmistuvan vuoden 2003 loppuun mennessä – vuoden 2004 puoliväliin mennessä, ja tuloksia sekä Arabidopsis-genomin primäärirakennetta koskevien tietojen kanssa käytetään laajasti vertailussa. muiden kasvien genomiikka.
Kuitenkin vuoden 2002 alussa kaksi tutkimusryhmää - toinen Kiinasta, toinen Sveitsistä ja Yhdysvalloista - julkaisi tulokset täydellisestä (likimääräisestä) riisin genomin sekvensointiluonnoksesta, joka suoritettiin käyttämällä kokonaiskloonaustekniikkaa. Toisin kuin vaiheittaisessa (hierarkkisessa) tutkimuksessa, kokonaislähestymistapa perustuu koko genomisen DNA:n samanaikaiseen kloonaukseen johonkin virus- tai bakteerivektoreista ja merkittävän (valtava määrä keskikokoisille ja suurille genomeille) yksittäisiä klooneja, jotka sisältävät erilaisia DNA-segmentit. Näiden sekvensoitujen osien analyysin ja DNA:n identtisten terminaalisten osien päällekkäisyyden perusteella muodostuu contig - DNA-sekvenssien ketju, jotka on liitetty yhteen. Yleinen (kokonais) jatkumo on koko genomin tai ainakin yksittäisen kromosomin ensisijainen rakenne.
Tällaisessa kaavamaisessa esityksessä kokonaiskloonauksen strategia näyttää yksinkertaiselta. Itse asiassa se kohtaa vakavia vaikeuksia, jotka liittyvät tarpeeseen hankkia valtava määrä klooneja (on yleisesti hyväksytty, että tutkittava genomi tai sen alue on päällekkäinen kloonien kanssa vähintään 10 kertaa), valtava määrä sekvensointia ja erittäin monimutkainen. kloonien telakointityö, joka edellyttää bioinformatiikan asiantuntijoiden osallistumista. Vakava este täydelliselle kloonaukselle ovat erilaiset toistuvat DNA-segmentit, joiden määrä, kuten jo mainittiin, kasvaa jyrkästi genomin koon kasvaessa. Siksi kokonaissekvensoinnin strategiaa käytetään pääasiassa virusten ja mikro-organismien genomien tutkimuksessa, vaikka sitä onkin menestyksekkäästi käytetty monisoluisen organismin, Drosophilan, genomin tutkimiseen.
Tämän genomin kokonaissekvensoinnin tulokset "asetettiin" valtavalle joukolle tietoa sen kromosomi-, geeni- ja molekyylirakenteesta, jotka saatiin lähes 100 vuoden Drosophilan tutkimusjakson aikana. Ja silti, sekvensointiasteen suhteen, Drosophilan genomi (66% genomin kokonaiskoosta) on huomattavasti huonompi kuin Arabidopsis-genomi (92%) huolimatta niiden melko läheisestä koosta - 180 miljoonaa ja 125 miljoonaa emäsparia, vastaavasti. . Siksi on äskettäin ehdotettu nimeämistä sekateknologialle, jota käytettiin Drosophilan genomin sekvensointiin.
Riisin genomin sekvensoimiseksi edellä mainitut tutkimusryhmät ottivat sen kaksi alalajia, laajimmin viljeltyjä Aasian maissa, - Oriza saliva L. ssp indicaj Ja Oriza saliva L. sspjaponica. Heidän tutkimustuloksensa ovat monessa suhteessa samat, mutta poikkeavat monilta osin. Siten molempien ryhmien edustajat ilmoittivat saavuttaneensa noin 92-93 % genomin päällekkäisyydestä jatkuvien kanssa. On osoitettu, että noin 42 % riisin genomista edustaa lyhyitä DNA-toistoja, jotka koostuvat 20 emäsparista, ja suurin osa liikkuvista DNA-elementeistä (transposoneista) sijaitsee intergeenisillä alueilla. Tiedot riisin genomin koosta vaihtelevat kuitenkin merkittävästi.
Japanilaisella alalajilla genomin kooksi on määritetty 466 miljoonaa emäsparia ja Intian alalajilla 420 miljoonaa. Syy tähän eroon ei ole selvä. Se voi olla seurausta erilaisista metodologisista lähestymistavoista genomien ei-koodaavan osan koon määrittämisessä, eli se ei heijasta asioiden todellista tilaa. Mutta on mahdollista, että tutkittujen genomien koossa on 15 prosentin ero.
Toinen suuri ero paljastui löydettyjen geenien määrässä: japanilaisessa alalajissa 46 022 - 55 615 geeniä genomia kohden ja intialaisessa alalajissa 32 000 - 50 000. Syy tähän eroon ei ole selvä.
Saatujen tietojen epätäydellisyys ja epäjohdonmukaisuus mainitaan julkaistujen artikkeleiden kommenteissa. Tässä ilmaistaan myös toive, että puutteet riisin genomin tietämyksessä poistetaan vertaamalla "karkean sekvensoinnin" tietoja kansainvälisen riisin genomiprojektin osallistujien suorittaman yksityiskohtaisen, hierarkkisen sekvensoinnin tuloksiin.
VERTAILEVA JA TOIMINNALLINEN KASVIGENOMIIKKA
Saatu laaja tieto, josta puolet (kiinalaisen ryhmän tulokset) on julkisesti saatavilla, avaa epäilemättä laajat näkymät sekä riisin genomin tutkimukselle että kasvigenomiikan tutkimukselle yleensä. Arabidopsiksen ja riisin genomien ominaisuuksien vertailu osoitti sen suurin osa Arabidopsiksen genomista tunnistetuista geeneistä (jopa 80 %) löytyi myös riisin genomista, mutta noin puolella riisistä löydetyistä geeneistä ei ole vielä löydetty analogeja (ortologeja) Arabidopsiksen genomista. Samaan aikaan 98 % geeneistä, joiden primäärirakenne on todettu muille viljoille, löytyi riisin genomista.
Merkittävä (melkein kaksinkertainen) ero riisin ja Arabidopsiksen geenien lukumäärän välillä on hämmentävää. Samanaikaisesti kokonaissekvensoinnilla saatuja riisin genomin dekoodauksen luonnostietoja ei käytännössä verrata riisin genomin tutkimuksen laajoihin tuloksiin hierarkkisen kloonauksen ja sekvensoinnin menetelmällä, eli siihen, mikä on ei ole tehty Drosophilan genomin suhteen. Siksi on epäselvää, heijastaako Arabidopsiksen ja riisin geenien lukumäärän ero asioiden todellista tilaa vai johtuuko se metodologisten lähestymistapojen eroista.
Toisin kuin Arabidopsiksen genomissa, riisin genomin kaksoisgeeneistä ei anneta tietoja. On mahdollista, että niiden suhteellinen määrä voi olla suurempi riisissä kuin Arabidopsisissa. Tätä mahdollisuutta tukevat epäsuorasti tiedot riisin polyploidisten muotojen esiintymisestä. Lisää selvyyttä tähän asiaan voidaan odottaa, kun kansainvälinen riisigenomiprojekti on saatu päätökseen ja yksityiskohtainen kuva tämän genomin primääri-DNA-rakenteesta on saatu. Vakavia perusteita tällaiselle toivolle antaa se, että riisin genomin karkeaa sekvensointia koskevien töiden julkaisemisen jälkeen tämän genomin rakennetta koskevien julkaisujen määrä on lisääntynyt jyrkästi, erityisesti on ilmestynyt tietoa yksityiskohtaisesta sekvensoinnista. sen 1 ja 4 kromosomista.
Kasvien geenien lukumäärän ainakin likimääräinen tietäminen on perustavanlaatuista vertailevan kasvigenomiikan kannalta. Aluksi uskottiin, että koska kaikki kukkivat kasvit ovat fenotyyppisiltä ominaisuuksiltaan hyvin lähellä toisiaan, myös niiden genomien tulisi olla samanlaisia. Ja jos tutkimme Arabidopsiksen genomia, saamme tietoa useimmista muiden kasvien genomeista. Epäsuora vahvistus tälle olettamukselle on hiiren genomin sekvensoinnin tulokset, joka on yllättävän lähellä ihmisen genomia (noin 30 tuhatta geeniä, joista vain 1 tuhat osoittautui erilaiseksi).
Voidaan olettaa, että syy Arabidopsiksen ja riisin genomien välisiin eroihin piilee niiden kuulumisessa eri kasviluokkiin - kaksisirkkaisiin ja yksisirkkaisiin. Tämän asian selventämiseksi on erittäin toivottavaa tietää ainakin jonkin muun yksisirkkaisen kasvin karkea primaarirakenne. Todellisin ehdokas voisi olla maissi, jonka genomi on suunnilleen sama kuin ihmisen genomi, mutta silti paljon pienempi kuin muiden viljojen genomit. Maissin ravintoarvo tunnetaan hyvin.
Arabidopsiksen ja riisin genomien sekvensoinnin tuloksena saadusta valtavasta materiaalista on vähitellen tulossa perusta laajamittaiselle kasvien genomien tutkimukselle vertailevaa genomiikkaa käyttäen. Tällaisilla tutkimuksilla on yleinen biologinen merkitys, koska niiden avulla voidaan selvittää kasvin genomin kokonaisuuden ja yksittäisten kromosomien järjestäytymisen pääperiaatteet, tunnistaa geenien ja niiden säätelyalueiden rakenteen yhteiset piirteet ja ottaa huomioon kromosomin toiminnallisesti aktiivisen (geeni)osan ja erilaisten intergeenisten DNA-alueiden suhde, jotka eivät koodaa proteiineja. Vertailevasta genetiikasta on tulossa yhä tärkeämpää myös ihmisen toiminnallisen genomiikan kehittämisessä. Pufferfish- ja hiiren genomien sekvensointi suoritettiin vertailevia tutkimuksia varten.
Yhtä tärkeää on tutkia yksittäisiä geenejä, jotka vastaavat yksittäisten proteiinien synteesistä, jotka määrittävät kehon tietyt toiminnot. Ihmisgenomiohjelman käytännön, ensisijaisesti lääketieteellinen merkitys piilee yksittäisten geenien löytämisessä, eristämisessä, sekvensoinnissa ja toiminnan määrittämisessä. Tämän seikan pani merkille useita vuosia sitten J. Watson, joka korosti, että Human Genome -ohjelma saataisiin päätökseen vasta, kun kaikkien ihmisen geenien toiminnot selvitetään.
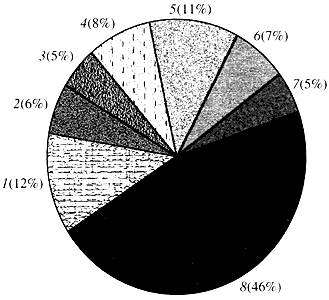
Riisi. 4. Luokittelu Arabidopsis-geenien toiminnan mukaan
1 - geenit kasvua, jakautumista ja DNA-synteesiä varten; 2 - RNA-synteesigeenit (transkriptio); 3 - geenit proteiinien synteesiä ja modifiointia varten; 4 - geenit kehitystä, ikääntymistä ja solukuolemaa varten; 5 - solujen aineenvaihdunnan ja energia-aineenvaihdunnan geenit; 6 - solujen välisen vuorovaikutuksen ja signaalinsiirron geenit; 7 - geenit muiden soluprosessien aikaansaamiseksi; 8 - geenit, joilla on tuntematon toimintaMitä tulee kasvigeenien toimintaan, tiedämme alle kymmenesosan siitä, mitä tiedämme ihmisen geeneistä. Jopa Arabidopsiksessa, jonka genomia on tutkittu paljon enemmän kuin ihmisen genomia, lähes puolet sen geeneistä on tuntematon (kuva 4). Samaan aikaan kasveilla on eläimille yhteisten geenien lisäksi huomattava määrä vain (tai ainakin pääosin) niille spesifisiä geenejä. Puhumme geeneistä, jotka osallistuvat veden kuljetukseen ja eläimillä puuttuvan soluseinän synteesiin, geeneistä, jotka varmistavat kloroplastien muodostumisen ja toiminnan, fotosynteesin, typen sitoutumisen sekä lukuisten aromaattisten tuotteiden synteesin. Tätä listaa voidaan jatkaa, mutta jo nyt on selvää, kuinka vaikea tehtävä kasvien toiminnallisella genomiikalla on edessään.
Koko genomin sekvensointi antaa lähes todellista tietoa tietyn organismin geenien kokonaismäärästä, mahdollistaa enemmän tai vähemmän yksityiskohtaisen ja luotettavan tiedon sijoittamisen niiden rakenteesta tietopankkeihin sekä helpottaa yksittäisten geenien eristämistä ja tutkimista. Genomisekvensointi ei kuitenkaan suinkaan tarkoita kaikkien geenien toiminnan vahvistamista.
Yksi toiminnallisen genomiikan lupaavimmista lähestymistavoista perustuu toimivien geenien tunnistamiseen, joita käytetään mRNA:n transkriptioon (lukemiseen). Tämä lähestymistapa, mukaan lukien moderni teknologia mikrosirujen avulla voit tunnistaa samanaikaisesti jopa kymmeniä tuhansia toimivia geenejä. Äskettäin tätä lähestymistapaa käyttäen on aloitettu kasvien genomien tutkimus. Arabidopsikselle oli mahdollista saada noin 26 tuhatta yksittäistä transkriptiota, mikä helpottaa suuresti mahdollisuutta määrittää melkein kaikkien sen geenien toiminta. Perunoista pystyttiin tunnistamaan noin 20 000 toimivaa geeniä, jotka ovat tärkeitä sekä kasvu- ja mukuloiden muodostumisprosessien että perunatautiprosessien ymmärtämiselle. Tämän tiedon oletetaan lisäävän yhden tärkeimmistä elintarvikkeista vastustuskykyä taudinaiheuttajia vastaan.
Funktionaalisen genomiikan looginen kehitys oli proteomiikkaa. Tämä uusi tieteenala tutkii proteomia, joka yleensä ymmärretään täydellisenä proteiinisarjana solussa tietyllä hetkellä. Tällainen genomin toiminnallista tilaa heijastava proteiinisarja muuttuu koko ajan, kun taas genomi pysyy muuttumattomana.
Proteiinien tutkimusta on käytetty pitkään arvioimaan kasvien genomien aktiivisuutta. Kuten tiedetään, kaikissa kasveissa esiintyvät entsyymit eroavat toisistaan yksittäisten lajien ja lajikkeiden osalta aminohappojen sekvenssissä. Tällaisia entsyymejä, joilla on sama toiminto, mutta erilainen yksittäisten aminohappojen sekvenssi, kutsutaan isoentsyymeiksi. Niillä on erilaisia fysikaalis-kemiallisia ja immunologisia ominaisuuksia ( molekyylimassa, varaus), joka voidaan havaita kromatografialla tai elektroforeesilla. Näitä menetelmiä on useiden vuosien ajan käytetty menestyksekkäästi tutkittaessa ns. geneettistä polymorfismia eli organismien, lajikkeiden, populaatioiden, lajien, erityisesti vehnän ja siihen liittyvien viljamuotojen välisiä eroja. Kuitenkin sisään Viime aikoina DNA-analyysimenetelmien, mukaan lukien sekvensoinnin, nopean kehityksen vuoksi proteiinipolymorfismin tutkiminen on korvattu DNA-polymorfismin tutkimuksella. Viljan tärkeimmät ravitsemukselliset ominaisuudet määrittävien varastoproteiinien (prolamiinit, gliadiinit jne.) spektrien suora tutkimus on kuitenkin edelleen tärkeä ja luotettava menetelmä maatalouskasvien geneettiseen analyysiin, valintaan ja siementuotantoon.
Geenien, niiden ilmentymis- ja säätelymekanismien tuntemus on erittäin tärkeää biotekniikan kehittämisen ja siirtogeenisten kasvien tuotannon kannalta. Tiedetään, että vaikuttavat menestykset tällä alalla aiheuttavat moniselitteisen reaktion ympäristö- ja lääketieteellisessä yhteisössä. Kasvibiotekniikassa on kuitenkin alue, jolla nämä pelot, elleivät täysin perusteettomia, näyttävät joka tapauksessa olevan vähäisiä. Puhumme siirtogeenisten teollisuuslaitosten luomisesta, joita ei käytetä elintarviketuotteina. Intiassa korjattiin äskettäin ensimmäinen sato siirtogeenistä puuvillaa, joka on vastustuskykyinen useille taudeille. Tietoa on erityisten pigmenttiproteiineja koodaavien geenien tuomisesta puuvillan genomiin ja sellaisten puuvillakuitujen tuotannosta, jotka eivät vaadi keinotekoista värjäystä. muu tekninen kulttuuri, joka voi olla tehokkaan geenitekniikan kohteena, on pellava. Sen käytöstä vaihtoehtona puuvillalle tekstiilien raaka-aineissa on keskusteltu viime aikoina. Tämä ongelma on erittäin tärkeä maallemme, joka on menettänyt omat raakapuuvillan lähteensä.
NÄKYMÄT KASVIGENOMIEN TUTKIMUKSESTA
On selvää, että kasvien genomien rakennetutkimukset perustuvat vertailevan genomiikan lähestymistapoihin ja menetelmiin, joissa päämateriaalina käytetään Arabidopsiksen ja riisin genomien purkamisen tuloksia. Tärkeä rooli vertailevan kasvigenomiikan kehittämisessä tulee epäilemättä olemaan tiedolla, joka ennemmin tai myöhemmin saadaan muiden kasvien genomien totaalisella (karkealla) sekvensoinnilla. Tässä tapauksessa vertaileva kasvigenomiikka perustuu geneettisten suhteiden selvittämiseen yksittäisten lokusten ja eri genomeihin kuuluvien kromosomien välillä. Emme keskity niinkään kasvien yleiseen genomiikkaan kuin yksittäisten kromosomaalisten lokusten selektiiviseen genomiikkaan. Esimerkiksi hiljattain on osoitettu, että vernaalisaatiosta vastaava geeni sijaitsee heksaploidisen vehnäkromosomin 5A VRn-AI-lokuksessa ja riisin kromosomin 3 Hd-6-lokuksessa.
Näiden tutkimusten kehittäminen on voimakas sysäys monien toiminnallisesti tärkeiden kasvigeenien tunnistamiseen, eristämiseen ja sekvensointiin, erityisesti geenien, jotka vastaavat taudinkestävyydestä, kuivuuden kestävyydestä ja sopeutumiskyvystä erilaisiin kasvuolosuhteisiin. Yhä enemmän käytetään toiminnallista genomiikkaa, joka perustuu kasveissa toimivien geenien massadetektioon (seulomiseen).
Voimme ennakoida kromosomitekniikoiden, ensisijaisesti mikrodissektiomenetelmän, parantamista edelleen. Sen käyttö laajentaa dramaattisesti genomitutkimuksen mahdollisuuksia ilman suuria kustannuksia, kuten esimerkiksi genomin kokonaissekvensointia. Menetelmä lokalisoida yksittäisten geenien kasvien kromosomeihin hybridisaation avulla leviää edelleen. paikan päällä. Tällä hetkellä sen käyttöä rajoittaa valtava määrä toistuvia sekvenssejä kasvin genomissa ja mahdollisesti kasvien kromosomien rakenteellisen järjestyksen erityispiirteet.
Kromosomitekniikoista tulee lähitulevaisuudessa suuri merkitys kasvien evoluutiogenomiikan kannalta. Nämä suhteellisen edulliset tekniikat mahdollistavat nopean sisäisen ja interspesifisen vaihtelun arvioinnin, tetraploidisen ja heksaploidisen vehnän, ruisvehnän kompleksisten allopolyploidisten genomien tutkimisen; analysoida evoluutioprosesseja kromosomitasolla; tutkia synteettisten genomien muodostumista ja vieraan geneettisen materiaalin sisääntuloa (introgressiota); tunnistaa geneettisiä suhteita eri lajien yksittäisten kromosomien välillä.
Genomin karakterisoinnissa käytetään kasvien karyotyypin tutkimusta klassisilla sytogeneettisillä menetelmillä, molekyylibiologisella analyysillä ja tietokonetekniikalla rikastettuna. Tämä on erityisen tärkeää tutkittaessa karyotyypin stabiilisuutta ja vaihtelua ei vain yksittäisten organismien, vaan myös populaatioiden, lajikkeiden ja lajien tasolla. Lopuksi on vaikea kuvitella, kuinka kromosomien uudelleenjärjestelyjen (poikkeamat, sillat) lukumäärä ja spektrit voidaan arvioida ilman differentiaalivärjäysmenetelmiä. Tällaiset tutkimukset ovat erittäin lupaavia seurannan kannalta ympäristöön kasvin genomin tilan mukaan.
SISÄÄN moderni Venäjä kasvien genomien suora sekvensointi on epätodennäköistä. Tällainen suuria investointeja vaativa työ on nykyisen taloutemme vahvuuksien ulkopuolella. Samaan aikaan maailmantieteen hankkimat ja kansainvälisistä tietopankeista saatavilla olevat tiedot Arabidopsiksen ja riisin genomien rakenteesta riittävät kotimaisen kasvigenomiikan kehittämiseen. Voidaan ennakoida vertaileviin genomiikan lähestymistapoihin perustuvien kasvigenomitutkimusten laajentamista jalostuksen ja kasvinviljelyn erityisongelmien ratkaisemiseksi sekä erilaisten taloudellisesti merkittävien kasvilajien alkuperän tutkimiseksi.
Voidaan olettaa, että meidän budjetillemme varsin edullisia genomisia lähestymistapoja, kuten geneettistä tyypitystä (RELF, RAPD, AFLP-analyysit jne.), käytetään laajasti kotimaisessa jalostuksessa ja kasvinviljelyssä. DNA-polymorfismin suorien määritysmenetelmien rinnalla genetiikan ja kasvinjalostuksen ongelmien ratkaisemisessa käytetään proteiinipolymorfismin, ensisijaisesti viljan varastoproteiinien, tutkimukseen perustuvia lähestymistapoja. Kromosomiteknologiaa tullaan käyttämään laajalti. Ne ovat suhteellisen edullisia, niiden kehittäminen vaatii melko maltillisia investointeja. Kromosomitutkimusten alalla kotimainen tiede ei ole huonompi kuin maailma.
On korostettava, että tieteemme on antanut merkittävän panoksen kasvien genomiikan muodostumiseen ja kehitykseen [ , ].
Pääroolin näytteli N.I. Vavilov (1887-1943).
Molekyylibiologiassa ja kasvigenomiikassa uraauurtava panos A.N. Belozersky (1905-1972).
Kromosomitutkimusten alalla on huomioitava erinomaisen geneetiikan S.G. Navashin (1857-1930), joka löysi ensimmäisenä satelliittikromosomit kasveista ja osoitti, että on mahdollista erottaa yksittäiset kromosomit niiden morfologian ominaisuuksien mukaan.
Toinen klassikko Venäjän tiede G.A. Levitsky (1878-1942) kuvasi yksityiskohtaisesti rukiin, vehnän, ohran, herneiden ja sokerijuurikkaan kromosomit, otti käyttöön termin "karyotyyppi" tieteeseen ja kehitti sen opin.
Nykyaikaiset asiantuntijat, jotka luottavat maailmantieteen saavutuksiin, voivat antaa merkittävän panoksen kasvigenetiikan ja genomiikan jatkokehitykseen.
Kirjoittaja ilmaisee sydämelliset kiitoksensa akateemikolle Yu.P. Altukhov kriittisestä keskustelusta artikkelista ja arvokkaista neuvoista.Artikkelin kirjoittajan johtaman ryhmän työtä tuki Venäjän perustutkimussäätiö (apurahat 99-04-48832; 00-04-49036; 00-04-81086), Venäjän federaatio tuki tieteelliset koulut(avustukset nro 00-115-97833 ja NSh-1794.2003.4) ja ohjelmat Venäjän akatemia Tieteet "Molekulaariset geneettiset ja kromosomaaliset markkerit nykyaikaisten jalostus- ja siemententuotantomenetelmien kehittämisessä".
KIRJALLISUUS
1. Zelenin A.V., Badaeva E.D., Muravenko O.V. Johdatus kasvien genomiikkaan // Molekyylibiologia. 2001. V. 35. S. 339-348. 2. Kynä E. Bonanza kasvien genomiikalle // Tiede. 1998. V. 282. P. 652-654. 3. Plant genomics, Proc. Natl. Acad. sci. USA. 1998. V. 95. P. 1962-2032. 4. Kartelli N.A. jne. Genetiikka. Ensyklopedinen sanakirja. Minsk: Technologia, 1999. 5. Badaeva E.D., Friebe B., Gill B.S. 1996. Genomin erilaistuminen Aegilopsissa. 1. Erittäin toistuvien DNA-sekvenssien jakautuminen diploidisten lajien kromosomeissa, genomi. 1996. V. 39. S. 293-306. Kromosomianalyysin historia // Biol. kalvot. 2001. T. 18. S. 164-172.
täysin määritelty. Siksi työ sukkulamatojen genomin tulkitsemiseksi olisi tunnustettava erittäin onnistuneeksi.
Vielä suurempi menestys liittyy Drosophilan genomin dekoodaukseen, vain vuonna
2 kertaa pienempi kuin ihmisen DNA ja 20 kertaa suurempi kuin sukkulamatojen DNA. Huolimatta korkea tutkinto Drosophilan geneettisessä tutkimuksessa noin 10 % sen geeneistä oli tuntemattomia siihen asti. Mutta paradoksaalisin on se tosiasia, että Drosophilassa, joka on paljon paremmin organisoitunut kuin sukkulamadot, osoittautui olevan vähemmän geenejä kuin mikroskooppisella sukkulamatolla! Sitä on vaikea selittää nykyajan biologisista kannoista. Enemmän geenejä kuin Drosophilassa on myös ristikukkaisten heimoon kuuluvan kasvin - Arabidopsiksen - dekoodatussa genomissa, jota geneetikot käyttävät laajasti klassisena koekohteena.
Genomiprojektien kehitystä seurasi monien tieteen ja teknologian alueiden intensiivinen kehitys. Joten bioinformatiikka sai voimakkaan sysäyksen kehitykselleen. Valtavien tietomäärien tallentamiseen ja käsittelyyn luotiin uusi matemaattinen laite; on suunniteltu ennennäkemättömän tehoisia supertietokonejärjestelmiä; on kirjoitettu tuhansia ohjelmia, joiden avulla voidaan muutamassa minuutissa tehdä vertaileva analyysi eri tietolohkoista, syöttää päivittäin uusia tietoja tietokoneen tietokantoihin,
eri laboratorioissa eri puolilla maailmaa saatuja tietoja ja mukauttaa uutta tietoa aiemmin kerättyyn tietoon. Samalla kehitettiin järjestelmiä genomin eri elementtien tehokkaaseen eristämiseen ja automaattiseen sekvensointiin eli DNA-nukleotidisekvenssien määrittämiseen. Tältä pohjalta on suunniteltu tehokkaita robotteja, jotka nopeuttavat merkittävästi sekvensointia ja tekevät siitä halvempaa.
Genomiikan kehitys on puolestaan johtanut valtavan määrän uusien tosiasioiden löytämiseen. Monien niiden merkitystä ei ole vielä arvioitu
tulevaisuus. Mutta jo nyt on ilmeistä, että nämä löydöt johtavat monien teoreettisten näkemysten uudelleenarviointiin koskien sen syntymistä ja kehitystä. useita muotoja elämää maan päällä. Ne auttavat ymmärtämään paremmin yksittäisten solujen toiminnan taustalla olevia molekyylimekanismeja ja niiden vuorovaikutuksia; monien tähän asti tuntemattomien biokemiallisten syklien yksityiskohtainen purkaminen;
analyysi niiden yhteydestä perusfysiologisiin prosesseihin.
Siten on siirtymä rakenteellisesta genomiikasta
toimiva, mikä puolestaan luo edellytykset sille
tutkimusta molekyyliemäkset solun ja koko organismin toimintaan.
Jo kerättyä tietoa analysoidaan aikana
muutaman seuraavan vuosikymmenen aikana. Mutta jokainen seuraava askel
Suunta purkaa eri lajien genomien rakennetta, tuottaa uusia teknologioita, jotka helpottavat tiedonhankintaprosessia. Niin,
alempana organisoituneiden elävien olentojen geenien rakennetta ja toimintaa koskevien tietojen käyttö voi nopeuttaa hakua merkittävästi
syrjäyttää melko työlästä molekyylimenetelmiä etsi geenejä.
Tietyn lajin genomin rakenteen purkamisen tärkein seuraus on kyky tunnistaa kaikki sen geenit ja
vastaavasti transkriptoitujen RNA-molekyylien ja kaikkien sen proteiinien molekyylien tunnistaminen ja määrittäminen. Analogisesti genomin kanssa syntyivät käsitteet transkriptomi, joka yhdistää transkription tuloksena muodostuneen RNA-molekyylien poolin, ja proteomi, joka sisältää monia geenien koodaamia proteiineja. Siten genomiikka luo pohjan uusien tieteiden - proteomiikan ja - intensiiviselle kehitykselle transkriptomiikka. Proteomiikka käsittelee kunkin proteiinin rakenteen ja toiminnan tutkimusta; analyysi proteiinikoostumus solut; yksittäisen solun toiminnan molekyyliperustan määrittäminen, joka on
monien satojen proteiinien koordinoidun työn tulos, ja
organismin fenotyyppisen piirteen muodostumisen tutkimus,
joka on tulosta miljardien solujen koordinoidusta työstä.
Hyvin tärkeä biologisia prosesseja esiintyy RNA-tasolla. Niiden analyysi on transkriptomiikan aihe.
Genomiikan alalla työskentelevien monien maailman maiden tutkijoiden suurimmat ponnistelut kohdistuivat kansainvälisen "Human Genome" -projektin ratkaisemiseen. Huomattava edistys tällä alalla liittyy idean toteuttamiseen,
J. S. Venterin ehdottama haku ja analysointi
ekspressoituja DNA-sekvenssejä, joita voidaan myöhemmin käyttää eräänlaisina "leimoina" tai genomin tiettyjen osien markkereina. Toinen riippumaton ja yhtä hedelmällinen lähestymistapa omaksui Fr.:n johtaman ryhmän työskentelyn.
Collins. Se perustuu ihmisen perinnöllisten sairauksien geenien ensisijaiseen tunnistamiseen.
Ihmisen genomin rakenteen selvittäminen johti sensaatiomaiseen löytöyn. Kävi ilmi, että ihmisen genomi sisältää vain 32 000 geeniä, mikä on useita kertoja vähemmän kuin proteiinien määrä. Samaan aikaan proteiinia koodaavia geenejä on vain 24 000, muiden geenien tuotteet ovat RNA-molekyylejä.
DNA-nukleotidisekvenssien samankaltaisuusprosentti eri yksilöiden, etnisten ryhmien ja rotujen välillä on 99,9 %.
Tämä samankaltaisuus tekee meistä ihmisiä - Homo sapiens! Kaikki vaihtelumme nukleotiditasolla mahtuu hyvin vaatimattomaan lukuun - 0,1 %.
Siten genetiikka ei jätä tilaa ajatuksille kansallisesta tai rodullisesta paremmuudesta.
Mutta katsokaa toisiamme - olemme kaikki erilaisia. Kansalliset ja varsinkin rodulliset erot ovat vieläkin havaittavissa. Kuinka monta mutaatiota määrää henkilön vaihtelevuuden ei prosentteina, vaan absoluuttisesti? Tämän arvion saamiseksi sinun on muistettava, mikä genomin koko on. Ihmisen DNA-molekyylin pituus on
3,2x109 emäsparia. 0,1 % tästä on 3,2 miljoonaa nukleotidia. Mutta muista, että genomin koodaava osa vie alle 3 % DNA-molekyylin kokonaispituudesta, ja tämän alueen ulkopuolella olevilla mutaatioilla ei useimmiten ole mitään vaikutusta fenotyyppiseen vaihteluun. Siten saadaksesi kokonaisarvion fenotyyppiin vaikuttavien mutaatioiden määrästä, sinun on otettava 3 % 3,2 miljoonasta nukleotidistä, mikä antaa meille luvun luokkaa 100 000. Eli noin 100 tuhatta mutaatiota muodostaa fenotyyppimme vaihtelua. Jos vertaamme tätä lukua geenien kokonaismäärään, käy ilmi, että geeniä kohden on keskimäärin 3-4 mutaatiota.
Mitä nämä mutaatiot ovat? Suurin osa (vähintään 70 %)
määrittää yksilöllisen ei-patologisen vaihtelumme, mikä erottaa meidät, mutta ei tee meistä huonompia toisiimme nähden. Tämä sisältää ominaisuuksia, kuten silmät, hiukset, ihon väri, vartalotyyppi, pituus, paino,
käyttäytymisen tyyppi, joka on myös suurelta osin geneettisesti määrätty, ja paljon muuta. Noin 5 % mutaatioista liittyy monogeenisiin sairauksiin. Noin neljännes jäljellä olevista mutaatioista kuuluu funktionaalisten polymorfismien luokkaan. Ne osallistuvat perinnöllisen alttiuden muodostumiseen laajalle levinneelle monitekijäiselle patologialle. Nämä arviot ovat tietysti melko karkeita.
mutta niiden avulla voimme arvioida ihmisen perinnöllisen vaihtelevuuden rakennetta.
Luku 1.16. Evoluution molekyyligeneettiset perusteet
Molekyylibiologian vallankumous, joka tapahtui vuosituhannen vaihteessa, huipentui monien satojen mikro-organismilajien genomien rakenteen purkamiseen sekä joidenkin alkueläintyyppien,
hiiva, kasvit, eläimet ja ihmiset, muuttivat monia klassisen genetiikan perinteisiä ajatuksia ja toivat lähemmäksi mahdollisuutta tutkia evoluution ja lajittelun molekyylimekanismeja. Syntyi uusi tiede - vertaileva genomiikka,
joka mahdollistaa yksittäisten molekyylien tasolla tapahtuvien evoluutionaalisesti merkittävien tapahtumien esiintymisen eri filogeneettisissä linjoissa. Kävi ilmi, että yleisessä tapauksessa evoluution edistyminen ei liity pelkästään, eikä niinkään geenien rakenteellisen organisaation määrän, pituuden ja jopa monimutkaisuuden lisääntymiseen, vaan paljon suuremmassa määrin geenien muutokseen. heidän työnsä säätely, joka määrää kymmenien tuhansien geenien koordinaation ja kudosspesifisen ilmentymisen. Viime kädessä tämä johti monimutkaisempien, erittäin spesifisten, monifunktionaalisten vuorovaikutteisten proteiinien kompleksien ilmestymiseen korkeampiin organismeihin, jotka pystyvät suorittamaan täysin uusia tehtäviä.
Tarkastellaanpa evoluutioprosessissa tapahtuvien muutosten luonnetta kolmella informaatiotasolla: DNA - RNA - proteiini tai genomi - transkripti - proteomi. Yleisesti voidaan sanoa, että kun elämän organisoinnin monimutkaisuus lisääntyy, genomin koko kasvaa. Siten prokaryoottisen DNA:n koko ei ylitä 8x106 emäsparia, siitä tulee kaksi kertaa suurempi hiivassa ja alkueläimissä, 10-15 kertaa suurempi hyönteisissä ja nisäkkäissä kasvu saavuttaa 3 suuruusluokkaa, eli tuhat kertaa ( 103).
Tämä suhde ei kuitenkaan ole lineaarinen. Joten nisäkkäissä emme enää näe merkittävää genomin koon kasvua. Lisäksi genomin koon ja elämän organisoinnin monimutkaisuuden välistä suhdetta ei aina ole mahdollista havaita. Siten joissakin kasveissa genomin koko on suuruusluokkaa tai jopa kaksi suuruusluokkaa suurempi kuin ihmisillä. Muista, että eukaryoottisen genomin koon kasvu verrattuna prokaryootteihin johtuu pääasiassa ei-koodaavien sekvenssien, toisin sanoen valinnaisten elementtien, ilmestymisestä. Olemme jo sanoneet, että ihmisen genomissa eksoneja on yhteensä enintään 1-3%. Ja tämä tarkoittaa, että geenien määrä korkeammissa organismeissa voi olla vain useita kertoja suurempi kuin mikro-organismeissa.
Eukaryoottiorganisaation monimutkaistuminen johtuu osittain siitä, että on syntynyt lisäsääntelyjärjestelmä, jota tarvitaan
varmistetaan kudosspesifinen geeniekspressio. Yksi eukaryooteissa syntyneiden geenien epäjatkuvan järjestäytymisen seurauksista oli vaihtoehtoisen silmukoinnin ja vaihtoehtoisen transkription laaja käyttö. Tämä johti uuden ominaisuuden syntymiseen valtavassa määrässä geenejä - kykyyn koodata useita toiminnallisesti erilaisia proteiini-isoformeja. Näin ollen proteiinien kokonaismäärä
eli proteomin koon, korkeammissa voi olla useita kertoja enemmän geenejä.
Prokaryooteissa geenien lukumäärän spesifinen vaihtelu on hyväksyttävää, ja
samankaltaiset erot monien mikro-organismien eri kantojen välillä
patogeenit mukaan lukien, voi olla kymmeniä prosentteja. Samanaikaisesti erilaisten mikro-organismien organisoinnin monimutkaisuus korreloi suoraan koodaavien sekvenssien lukumäärän ja pituuden kanssa.
Siten fenotyypin sisäinen ja lajien välinen vaihtelu on tiukasti yhteydessä transkription ja proteomin kokoihin, jotka ovat arvoltaan hyvin samanlaisia. Eukaryooteissa geenien lukumäärä on jäykästi määrätty lajin ominaisuus, ja evoluution monimutkaisuuden lisääntyminen perustuu eri periaatteeseen - rajoitetun ja melko vakaan proteomin eri komponenttien differentiaaliseen monitasoiseen käyttöön.
Sukkulamato- ja Drosophila-genomien sekvensointi on osoittanut, että proteomikoot näissä hyvin erilaisissa lajeissa ovat hyvin läheisiä ja vain kaksi kertaa suurempia kuin hiivassa ja joissakin bakteerilajeissa. Tämä säännöllisyys – eri elämänmuotojen järjestäytymisen monimutkaisuuden merkittävä lisääntyminen samalla kun proteomin koko säilyy tai suhteellisen vähän kasvaa – on ominaista kaikelle myöhemmälle evoluutiolle aina ihmiseen asti. Niin,
ihmisen ja hiiren proteomit eivät käytännössä eroa toisistaan ja ovat alle 2 kertaa suurempia kuin mikroskooppisen sukkulamatomadon tai Drosophila-hedelmäkärpäsen proteomit. Lisäksi ihmisen DNA:n nukleotidisekvenssien identiteetti ja
suuria afrikkalaisia apinoita on 98,5%, ja koodausalueilla saavuttaa 99%. Nämä luvut poikkeavat vähän 99,9 prosentin arvosta.
DNA:n nukleotidisekvenssien sisäisen samankaltaisuuden määrittäminen planeetallamme asuvien eri yksilöiden, kansojen ja rotujen välillä. Mitkä ovat siis tärkeimmät muutokset, jotka muodostavat enintään 1,5 % koko genomista ihmisen muodostumisen kannalta? Ilmeisesti vastausta tähän kysymykseen ei pitäisi etsiä vain genomiselta ja proteomiselta tasolta.
Todellakin, yhdessä proteomin suhteellisen stabiilisuuden kanssa
Evoluution aikana eukaryoottisen transkription organisaation koko ja monimutkaisuus kasvavat jyrkästi, koska genomiin ilmestyy valtava määrä transkriptoitua ja ei-koodaavaa DNA:ta sekä merkittävästä laajenemisesta. RNA:ta koodaavien geenien luokka. RNA:t, jotka eivät koodaa proteiineja, joiden päälähde ovat intronit,
muodostavat suurimman osan korkeampien organismien transkriptistä,
saavuttaen 97-98 % kaikista transkriptioyksiköistä. Tällä hetkellä näiden molekyylien toimintoja analysoidaan intensiivisesti.
Siten tärkeimmät evoluutiomuutokset tapahtuvat genomin koon kasvun, melko vakaan proteomin ja transkription koon jyrkän kasvun taustalla (kuva 1). 31.
Kuva 31. Evoluutiomuutokset tapahtuvat kolmella
tietotasot Samaan aikaan siirtyminen yksinkertaisista elämänmuodoista monimutkaisempiin on ilmeistä
korreloi kahden perustavanlaatuisen ja jossain määrin toisiinsa liittyvän evolutionaarisen hankinnan syntymisen ja laajan leviämisen kanssa genomissa: ei-koodaavan DNA:n ja toistuvien elementtien. Suora seuraus näistä genomisella tasolla tapahtuvista muutoksista on valtavan määrän RNA:ita, jotka eivät koodaa proteiineja, ilmaantuminen evoluutioprosessiin.
Mikä on näiden evolutionaaristen muutosten rakenteellinen perusta?
Kaikkiin tärkeimpiin evolutionaarisiin siirtymisiin: prokaryooteista eukaryooteihin, alkueläimistä monisoluisiin organismeihin, ensimmäisistä eläimistä bilateraalisiin organismeihin ja primitiivisistä sonnesoluista selkärankaisiin liittyi genomin monimutkaisuuden voimakas lisääntyminen. Ilmeisesti tällaiset evoluutiohypyt ovat seurausta harvoista tapauksista, joissa on onnistuttu fuusioimaan eri lajien kokonaisia genomeja, jotka kuuluvat systemaattisiin luokkiin, jotka erosivat huomattavan etäisyyden päässä toisistaan. Siten arkeoiden ja bakteerien symbioosi merkitsi siirtymisen alkua prokaryooteista eukaryooteihin. Ilmeisesti mitokondriot, kloroplastit ja jotkut muut soluorganellit ilmestyivät myös endosymbioosin seurauksena. Korkeampien eukaryoottien perusominaisuus, diploidia, syntyi noin 500 miljoonaa vuotta sitten tapahtuneesta hyvin säädellystä genomisen kaksinkertaisuudesta.
Genomisen päällekkäisyyksiä lajin sisällä tapahtui melko usein ja
esimerkkejä tästä ovat lukuisat polyploidiatapaukset kasveissa,
sieniä ja joskus jopa eläimiä. Kuitenkin mahdollisia mekanismeja
Pohjimmiltaan uusien elämänmuotojen syntymiseen evoluutioprosessissa ei johdu autopolyploidia, vaan hybridisaatio ja genomien horisontaalinen siirto tai fuusio. On huomionarvoista, että merkittävimmät evoluutiomuutokset, joihin liittyy kokonaisten genomien fuusio, tapahtuvat poikkeuksellisissa olosuhteissa, suurten geologisten muutosten aikoina, kuten ilmakehän happipitoisuuden muutoksissa, Maan jäätyessä tai kambrian räjähdyksessä.
Suhteellisen rauhallisissa geologisissa olosuhteissa yksittäisten geenien tai kromosomisegmenttien päällekkäisyydet ja niiden myöhemmät erot osoittautuvat evoluution kannalta merkittävämmiksi. Sekvensoitujen genomien nukleotidisekvenssien vertailu osoittaa, että geenien kaksinkertaistumistiheys on melko korkea ja keskimäärin 0,01 geeniä kohti miljoonassa vuodessa. Suurin osa niistä ei ilmene seuraavien miljoonien vuosien aikana ja vain harvoissa tapauksissa
tapauksissa kaksinkertaiset geenit voivat saada uusia adaptiivisia toimintoja. Siitä huolimatta suuri joukko "hiljaisia" geenikopioita toimii eräänlaisena vararahastona uusien geenien syntymiselle ja uusien lajien muodostumiselle. Ihmisen genomi sisältää 10 000 - 20 000 kopiota prosessoituja geenejä, jotka ovat syntyneet mRNA:n retropositiosta.
Suurin osa niistä kuuluu pseudogeenien luokkaan, toisin sanoen niitä ei ekspressoidu joko mutaatioiden esiintymisen vuoksi tai johtuen insertiosta genomin transkriptionaalisesti inaktiivisille alueille. Jotkut näistä geeneistä ovat kuitenkin aktiivisia, ja niiden ilmentymisen luonne ja jopa toiminnot voivat olla erilaisia,
kuin perustaa geenejä.
Erityinen rooli kädellisten ja ihmisten evoluutiossa on segmenttien päällekkäisyyksiä jotka kuuluvat matalakopioisten toistojen luokkaan (LCR) ja
syntyi alle 35 miljoonaa vuotta sitten. Nämä sekvenssit ovat erittäin identtisiä DNA-lohkoja, joiden koko vaihtelee yhdestä useaan sataan kiloemäkseen. Useimmiten segmentaaliset päällekkäisyydet lokalisoituvat pericentromeerisille tai telomeerisille alueille. erilaisia kromosomeja, ja yhteensä ne vievät noin 5 % ihmisen genomista.
Muissa sekvensoiduissa genomeissa ei havaittu segmentaalisia päällekkäisyyksiä.
Segmentaalisen monistumisen pienin yksikkö, jota kutsutaan duplikoniksi, sisältää fragmentteja toisiinsa liittyvistä käsittelemättömistä geeneistä, ja
tämä erottaa sen muista tunnetuista toistuvien sekvenssien tyypeistä. Tietyissä olosuhteissa duplikonit voivat toimia lähteinä uusien kimeeristen transkriptoitujen geenien tai geeniperheiden luomiseen niissä esiintyvien koodaavien eksonien erilaisista yhdistelmistä. Joidenkin arvioiden mukaan 150-350 geeniä voi erottaa simpanssin ja ihmisen genomin.
Pienentämättä uusien koodaavien sekvenssien ilmaantumista ja vanhojen koodaavien sekvenssien katoamista koskevien tosiasioiden tärkeyttä spesifioinnin kannalta, on korostettava muiden mekanismien olemassaolon todellista mahdollisuutta,
sillä on ratkaiseva rooli eukaryoottien kehityksessä.
Yksi evoluution liikkeelle panevista mekanismeista ovat liikkuvia elementtejä, joita löytyy kaikista tässä suhteessa tutkituista lajeista.
Spesiaatioprosessiin liittyvät genomin muutokset voivat sisältää laajoja karyotyyppien uudelleenjärjestelyjä, paikallisia kromosomien uudelleenjärjestelyjä, geeniperheiden päällekkäisyyksiä, yksittäisten geenien modifikaatioita,
niihin liittyy niiden syntymä tai menetys, sekä erot geenien ilmentymisessä, joita säädellään sekä transkription että silmukoinnin tai translaation tasolla. Mobiilielementit liittyvät suoraan kaikkiin näihin prosesseihin.
Joissakin tapauksissa transposoitavissa elementeissä itsellään on sekvenssejä, jotka koodaavat entsyymejä, joiden läsnäolo on välttämätöntä DNA:n transponoinnin tai RNA:n retroposition aikaansaamiseksi.
Samanlaisia sekvenssejä on retrovirusten genomissa, LTR-
elementtejä ja transposoneja. Lukuisin transposoituvien elementtien luokka, Alu-repeats, kuuluu myös retrotransposonien ryhmään. Ensimmäistä kertaa Alu-
toistoja esiintyy kädellisissä noin 50-60 miljoonaa vuotta sitten pienestä RNA:ta koodaavasta geenistä. Jatkokehityksen prosessissa tämän perheen eroaminen ja voimakas vahvistuminen tapahtuu. Kädellisistä ihmisiin siirtymiseen liittyy lukumäärän räjähdysmäinen kasvu
Alu-toistot, joiden kopiomäärä joidenkin arvioiden mukaan yltää
1,1 miljoonaa. Alu-toistot vievät noin 10 % ihmisen genomista, mutta niiden jakautuminen on epätasaista, koska ne liittyvät enemmän geeneihin. Näitä elementtejä esiintyy harvoin koodaavissa eksoneissa ja niitä löytyy usein mRNA:n introneista ja ei-koodaavista alueista vaikuttamaan näiden molekyylien stabiilisuuteen ja/tai translaation tehokkuuteen. Alu-sekvenssien esiintymiseen geenien intronialueilla voi liittyä muutos preRNA-käsittelyn luonteessa, koska nämä sekvenssit sisältävät alueita, jotka ovat homologisia luovuttajan ja vastaanottajan silmukointikohtien kanssa. Alu-elementtien liittäminen geenin säätelyalueille voi häiritä transkriptiota, mikä johtaa
© M.D. Golubovsky
Ei-kanoniset perinnölliset muutokset
M.D. Golubovski
Mihail Davidovich Golubovski, lääkäri biologiset tieteet, johtava tutkija
Venäjän tiedeakatemian luonnontieteen ja tekniikan historian instituutin Pietarin haara.
Genetiikka tieteenä muotoutui 100 vuotta sitten Mendelin lakien toisen löytämisen jälkeen. Sen nopeaa kehitystä leimasi viime vuosina monien kymmenien lajien genomin DNA:n nukleotidikoostumuksen purkaminen. Syntyi uusia tietämyksen aloja - genomiikka, molekyylipaleogenetiikka. Vuoden 2001 alussa osana kallista 10 vuotta kansainvälinen ohjelma ilmoitti ihmisen genomin perustavanlaatuisen dekoodauksen. Näitä saavutuksia voidaan ehkä verrata miehen avaruuskävelyyn ja kuuhun laskeutumiseen.
Geenitekniikka ja biotekniikka ovat muuttaneet tieteen kasvot suuresti. Tässä on utelias jakso, joka sisältyy jo viimeisimpään yhteenvetoon: "Vuoden 1998 jälkeen alkoi ennennäkemätön kilpailu maailmanlaajuisen Human Genome Project -yhteisön 1 100 tutkijan ja pääomasijoitusyhtiö Celera Genomicsin välillä". Yritys toivoi ylittävänsä ensimmäisenä maaliviivan ja hyötyvänsä ihmisen DNA-fragmenttien patentoinnista. Mutta toistaiseksi periaate on voittanut: "Ihminen ei voi patentoida sitä, mikä on luonnon ja Jumalan luomaa."
Voisiko Gregor Mendel kuvitella niin fantasmagorisen kuvan, kun hän vietti hitaasti kokeitaan vuosi toisensa jälkeen luostarin puutarhan hiljaisuudessa? Missä määrin se muuttaa tieteen luonnollista itsekehitystä? Poistaako genomien täydellinen DNA-analyysi todella kaikki kannet? Toivoo, että Pinocchio olisi jo löytänyt arvokkaan kultaisen avaimen salaisen oven edessä odottamattoman todellisuuden ja paradoksien kanssa. Ihmisillä vain 3 % genomin DNA:sta koodaa proteiineja, ja ehkä vielä 20-25 % osallistuu geenien toiminnan säätelyyn. Mikä on tehtävä, ja onko muulla DNA:lla se? Genomin geenejä verrataan joskus pieniin saariin inaktiivisten ja mahdollisesti roskaisten sekvenssien meressä. DNA-rotu muistuttaa joskus sanontaa: "Tuo se, en tiedä mitä."
Skeptikoiden vastalauseita ei ole millään tavalla poistettu. Todellakin, täydellisellä sekvensoinnilla tietyn DNA-segmentin nimeäminen (käytän muodikasta termiä) "geeniluokissa" suoritetaan vain puhtaasti muodollisten kriteerien perusteella (transkriptiota varten välttämättömät geneettiset välimerkit). Useimpien "nimettyjen geenien" rooli, aika ja toimintapaikka ovat vielä täysin epäselviä.
Mutta on toinenkin ongelma. Genomi tulee ymmärtää koko perinnöllisenä järjestelmänä, joka sisältää paitsi tietyn DNA-elementtijoukon rakenteen, myös niiden välisten yhteyksien luonteen, joka määrää ontogeneesin kulun tietyissä ympäristöolosuhteissa. On olemassa systeeminen kolmikko: elementit, niiden väliset yhteydet ja eheyden ominaisuudet. Tästä seuraa tärkeä johtopäätös: geenien rakenteen tuntemus DNA-tasolla on välttämätöntä, mutta ei ollenkaan riittävää genomin kuvaamiseen. Olemme vasta dynaamisen organisointitavan ja ei-kanonisten periytymismuotojen ymmärtämisen kynnyksellä [ , ].
Yllättäen 1900-luvun lopulla. kysymys siitä, mitkä ovat perinnöllisen vaihtelevuuden rajat ja kirjo, on mennyt puhtaasti akateemisen keskustelun ulkopuolelle. Ensin Englannissa ja sitten Saksassa karja jouduttiin teurastamaan hermostoa rappeuttavan poikkeaman vuoksi, joka voi tarttua ihmisiin sairaiden eläinten lihan mukana. Tartunnan aiheuttaja ei osoittautunut DNA:ta tai RNA:ta, vaan proteiineja, joita kutsutaan prioneiksi (englannin kielestä prions - protein infectious particles - protein infectious particles).
Ensimmäistä kertaa tutkijat kohtasivat heidän epätavallisen ilmenemismuotonsa jo 60-luvulla. Mutta sitten he yrittivät tulkita tätä ilmiötä klassisten käsitteiden puitteissa uskoen, että nämä olivat eläinten "hitaita virusinfektioita" tai erityisiä suppressorimutaatioita hiivassa. Nyt se käy ilmi "Prioniilmiö ei ole nisäkkäiden eksoottinen ominaisuus, vaan pikemminkin yleisen biologisen mekanismin erikoistapaus" dynaaminen perintö. Todennäköisesti molekyyligenetiikan keskeistä dogmaa joudutaan täydentämään ottaen huomioon infektiotyypin sisäisen ja interspesifisen leviämisen mahdollisuus.
80-luvun alussa molekyylibiologian ja genetiikan klassikko R.B. Khesin tunnisti kolme ei-kanonisen perinnöllisen vaihtelevuuden muotoa: ei-satunnaiset muutokset DNA-toistoista koostuvien kromosomien lokuksissa ja alueilla; sytoplasman ominaisuuksien muutos ja periytyminen; kromatiinipakkauksen paikallisten ja yleisten muutosten epigeneettinen periytyminen. Sitten lisättiin liikkuvia geenejä, joiden käyttäytyminen johti genomin epäjohdonmukaisuuden ongelmaan.
Tämän artikkelin tarkoitus on osoittaa se erilaisia muotoja Ei-Mendelin perintö ei ole poikkeus, vaan seuraus muusta yleisiä ideoita genomin järjestäytymisestä. Perinnölliset muutokset eivät suinkaan rajoitu mutaatioihin.
Andre Lvov ja hänen löytönsä rooli
Hämmästyttävän sattuman seurauksena samassa vuonna 1953 ilmestyi kaksi artikkelia, jotka määrittelivät kasvot nykyaikainen genetiikka: J. Watsonin ja F. Crickin löytämä DNA:n kaksoiskierteen ja A. Lvovin (1902-1994) käsite bakteerien profaagin ja lysogeniasta, joka on mielestäni nyt yhtä tärkeä biologian kannalta, lääketiede ja genetiikka kuin kaksoiskierre DNA.
Lvov totesi, että faagi voidaan integroida bakteerin kromosomiin ja siirtyä useiden sukupolvien ajan normaalin bakteerigeenin tavoin. Tässä tilassa faagissa toimii vain repressorigeeni, joka estää kaikkien sen muiden lokusten toiminnan. Bakteereja, jotka ovat sisällyttäneet faagin genomiinsa, kutsutaan lysogeeniseksi bakteeriksi, ja upotettua faagia kutsutaan profaagiksi. Tällainen lysogeeninen bakteeri on suojattu muiden faagien aiheuttamalta infektiolta. Ultraviolettisäteilyn tai solun sisäisen ympäristön muutosten vaikutuksesta repressori inaktivoituu, esto poistetaan ja faagi lisääntyy aiheuttaen solukuoleman. Nyt on jopa vaikea kuvitella, kuinka vallankumouksellinen tämä löytö oli.
Andre Lvov - kotoisin Venäjältä, hänen vanhempansa muuttivat Ranskaan 1800-luvun lopulla. Tiedemies Maria Siminovitšin äidin kuva on ikuisesti painettu taiteilija V. Serovin kankaalle "Auringon valaisema tyttö" (1888). Maria Yakovlevna Lvova-Siminovich eli 90-vuotiaaksi. Muutama viikko ennen toista maailmansotaa hän lahjoitti V. Serovin kirjeitä ja piirustuksia Tretjakovin gallerialle. Lvovin isä tunsi Mechnikovin ja vei poikansa tapaamaan häntä Pasteur-instituuttiin. Siten kulttuurin langat venyvät ja kietoutuvat vuosisatojen ja maiden läpi. minun puolestani pitkä elämä A. Lvov työskenteli peräkkäin alkueläinlääkärinä, bakteriologina, biokemistinä, geneetikkona ja lopulta virologina. Pasteur-instituutissa hän holhosi sekä J. Monodia että F. Jacobia, jotka jakoivat vuoden 1965 Nobel-palkinnon mestarin kanssa operonin löytämisestä.
1920-luvulta lähtien on tiedetty bakteerikantoja, joiden väitetään kantavan faageja piilevässä tilassa ja aiheuttavan ajoittain solujen hajoamista. Bakteriofagin löytäjä F.D. "Errel kuitenkin katsoi faagia vain solulle tappavana aineena, eikä sallinut ajatusta sen piilevasta tilasta. Tämän mielipiteen yhtyi aluksi molekyyligenetiikan klassikko M. Delbrück. Tosiasia on, että että hän ja hänen kollegansa Yhdysvalloissa työskentelivät ns. T-faagien kanssa, jotka eivät pysty integroitumaan bakteerikromosomiin. "Auktoriteettidemonin" vuoksi lysogeniaa ei ole tutkittu tarkasti 1920-luvun jälkeen. Tässä työssä loistava mikrobiologi Pasteur-instituutista Eugene Wolman joutui saksalaisten vangiksi juutalaiseksi Pariisin miehityksen aikana ja kuoli.
Sodan jälkeen Lvov aloitti uudelleen piilevän faagipitoisuuden tutkimuksen Pasteur-instituutissa. Vuonna 1953 hän loi johdonmukaisen profagin käsitteen, ymmärtäen heti sen merkityksen syövän virusteorialle ja useille viruspatologioille ihmisillä. Hänen selkeä suunnitelmansa lysogenian ilmiöstä esitetään edelleen kaikissa molekyyligenetiikan tiivistelmissä.
Vuonna 1958 F. Jacob ja Elias Wolman (Eugene Wolmanin poika) ottivat käyttöön termin episomi elementeille, jotka voivat esiintyä joko vapaassa tilassa tai integroituneena isäntägenomiin. He kutsuivat episomeja lauhkeiksi faageiksi, bakteerien sukupuolitekijöiksi, kolisinogeenisuustekijöiksi, joiden avulla jotkut bakteerikannat tappavat muita bakteereja. Merkittävässä kirjassa "Bakteerien sukupuoli ja genetiikka", joka kirjoitettiin vuonna 1961 (ja joka julkaistiin venäjäksi tunnetun geneetikko S.I. Alikhanyanin toimesta ensi vuonna), kirjoittajat ennakoivat episomimaisten elementtien olemassaolon korkeammissa organismeissa. viittaavat "ohjaaviin elementteihin", jonka B. McClintock löysi 50-luvun alussa ( Nobel palkinto fysiologiassa tai lääketieteessä 1983). He eivät kuitenkaan tuolloin ymmärtäneet, kuinka syvä tämä analogia on. Sen jälkeen kun 1970-luvun alussa löydettiin insertiomutaatioita, jotka aiheutuivat virus-DNA:n liittämisestä bakteerien solugenomiin, tuli mahdolliseksi rakentaa evoluutiosarja kahdenvälisiä siirtymiä: faagien "plasmidien" "transposonien" insertiosegmenttejä.
Samanlaisia reinkarnaatioita löydettiin eukaryooteista. Drosophilassa mustalaisperheen liikkuvat elementit ("mustalaiset") voivat esiintyä kopioina, jotka on rakennettu kromosomiin; olla täydellisten tai pelkistettyjen ympyrämäisten tai lineaaristen plasmidien muodossa sytoplasmassa; Lopuksi, jos isäntägenomissa on yksittäisiä "sallivia" mutaatioita, ne pystyvät pukeutumaan kuoreen, niistä tulee todellisia tarttuvia retroviruksia ja infektoivat vieraita isäntiä ruoan kautta. Drosophilan P-transposonien ja ihmisten endogeenisen retroviruksen HIV:n samankaltaisuus (taulukko) mahdollistaa mahdollisten evoluutiogeneettisten tapahtumien ennustamisen ihmispopulaatioissa, sen väistämättömän kohtalon nyt ja tulevissa kontakteissa vieraiden genomien kanssa.
Fakultatiivinen periaate ja genomin yleinen käsite
Monet transponoitaviin elementteihin liittyvät vaihtelevuuden tosiasiat eivät sovi mutaatioiden käsitteeseen paikallisina muutoksina geenilokusten rakenteessa, lukumäärässä tai sijainnissa. Klassisen ja "liikkuvan" genetiikan tietojen yhdistämiseksi ehdotin vuonna 1985 genomielementtien luonnollista luokittelua, joka sisältää kaksi alajärjestelmää: obligaatit (geenit ja niiden säätelyalueet kromosomeissa) ja fakultatiiviset elementit (DNA- ja RNA-kantajat, lukumäärä ja joiden topografia vaihtelee saman lajin eri soluissa tai organismeissa).
Tästä luokittelusta seuraa tärkeitä seurauksia, joiden avulla on mahdollista ymmärtää tai muotoilla monia epätavallisia tosiasioita perinnöllisen vaihtelun alalta. Nimetään muutama niistä:
- valinnaisuuden monipuolisuus. Ei ole olemassa lajigenomeja, jotka koostuvat vain pakollisista elementeistä, kuten ei ole olemassa eläviä organismeja, jotka koostuvat vain luurangosta;
- tytärsolujen geneettinen epäidenttisyys. Sattumalta ne eroavat sytoplasmisten fakultatiivisten elementtien lukumäärästä ja koostumuksesta. Pakollisten ja fakultatiivisten DNA-elementtien fraktioiden suhde on suhteellisen vakaa lajin ominaisuus. Sukulaislajeissa, joilla on samanlainen määrä geenilokuksia, DNA:n määrä voi poiketa 2–5 kertaa tai useamminkin, mikä lisää toistolohkoja ja muuttaa niiden genomista topografiaa. Erilaisia siirtymiä havaitaan jatkuvasti genomin pakollisen ja fakultatiivisen osan välillä. Ilmeisimpiä esimerkkejä ovat geenimutaatiot, jotka johtuvat liikkuvien elementtien tuomisesta (lisäämisestä) tai kromosomisegmenttien lisääntymisestä (amplifikaatiosta) ja niiden siirtymisestä erilaisiin kromosomin sisäisiin ja ekstrakromosomaalisiin tiloihin;
- tyypillinen perinnöllinen vaihtelutyyppi genomin kummallekin alajärjestelmälle. Morganimutaatiot korreloivat helposti pakollisen komponentin kanssa. Ehdotin, että erilaisia perinnöllisiä muutoksia valinnaisten elementtien lukumäärässä ja topografiassa kutsuttaisiin "variaatioiksi" (kuten musiikissa - muunnelmia tietystä teemasta). Mutaatiot klassisten käsitteiden mukaan tapahtuvat yleensä sattumalta, alhaisella taajuudella yksittäisissä yksilöissä. Muunnelmien luonne on täysin erilainen - massiiviset, järjestetyt muutokset ovat mahdollisia täällä erilaisten, mukaan lukien heikkojen, ei-mutageenisten tekijöiden (lämpötila, ruoka-ohjelma jne.) vaikutuksesta;
- luonnollisten perinnöllisten muutosten kaksivaiheinen luonne. Ensinnäkin valinnaiset elementit aktivoidaan herkimmiksi ympäristön muutoksille. Sitten myös geenilokukset alkavat vaikuttaa epäsuorasti. Päädyimme tähän johtopäätökseen monivuotisten havaintojen aikana luonnossa esiintyvien mutaatioiden puhkeamisesta. Suurin osa niistä osoittautui epävakaiksi ja johtui liikkuvien elementtien lisäyksistä, jotka aktivoidaan mystisesti ajoittain luonnossa. Drosophilassa noin 70 % luonnossa tai laboratoriossa spontaanisti syntyneistä mutaatioista liittyy liikkuvien elementtien liikkeeseen.
Yleinen ajatus genomista pakollisten ja fakultatiivisten elementtien kokonaisuutena laajentaa myös "horisontaalisen siirron" käsitettä, joka ei sisällä vain vieraiden geenien integrointia ytimen kromosomeihin. Horisontaalisesta siirrosta voidaan puhua silloinkin, kun syntyy kahden geneettisen järjestelmän stabiili yhdistys, jossa ilmaantuu uusia piirteitä ja ominaisuuksia.
Genomin toiminnallinen valinnaisuus
Perinnölliset muutokset syntyvät prosessien virheiden seurauksena perinnöllinen materiaali kaikki elävät organismit - replikaatio, transkriptio, translaatio sekä korjaus ja rekombinaatio.
Fakultatiivisella replikaatiolla tarkoitetaan yksittäisten DNA-segmenttien suhteellisen autonomista hyper- tai hyporeplikaatiota riippumatta koko genomisen DNA:n suunnitellusta säännöllisestä replikaatiosta solunjakautumisen aikana. Tällaisia ominaisuuksia omaavat kromosomien osat, joissa on toistoja, heterokromatiinilohkoja. Tässä tapauksessa autonominen replikointi johtaa yksittäisten segmenttien lukumäärän kertomiseen, ja sillä on yleensä mukautuva luonne.
Transkription fakultatiivinen luonne koostuu mahdollisuudesta ilmaantua eri mRNA:ita samasta templaatista johtuen useamman kuin yhden promoottorin läsnäolosta ja vaihtoehtoisesta silmukoitumisesta tietyssä lokuksessa. Tämä tilanne on normaali monille geeneille.
Translaation monitulkintaisuus (S.G. Inge-Vechtomovin terminologiassa) ilmenee saman kodonin eri tunnistusmuunnelmissa, esimerkiksi lopetuskodonissa tai kodonissa tietyn aminohapon sisällyttämiseksi syntetisoituun proteiiniin. Tällainen translaatio riippuu solun fysiologisista olosuhteista ja genotyypistä.
M.E. Lobaševin mutaatioprosessin teorian mukaan mutaation esiintyminen liittyy solun ja sen perinnöllisten rakenteiden kykyyn korjata vaurioita. Tästä seuraa, että mutaation ilmaantumista edeltää tila, jolloin vaurio on joko täysin palautuva tai se voidaan toteuttaa mutaation muodossa, joka ymmärretään "ei-identtisenä korjauksena". 1970-luvun alussa kävi selväksi, että DNA:n stabiilius solussa ei ole itse DNA-molekyylien immanentti ominaisuus - sitä ylläpitää erityinen entsymaattinen järjestelmä.
1970-luvun puolivälistä lähtien "rekombinaatiovirheiden" evoluution rooli perinnöllisten muutosten indusoijana, joka on paljon voimakkaampi kuin DNA:n replikaatiovirheet, alkoi tulla selväksi.
Molekyylitasolla on kolme tyyppiä rekombinaatiota: yleinen, paikkaspesifinen ja replikatiivinen. Ensimmäisessä, yleisessä, säännöllisessä rekombinaatiossa (crossing over) korjaus sisältää DNA-ketjun katkeamisen, niiden silloittumisen ja korjauksen. Se vaatii pitkiä DNA-homologian alueita. Kohdespesifinen rekombinaatio sisältää lyhyitä, useita emäksiä, homologia-alueita, joissa on esimerkiksi faagin 1 DNA ja bakteerikromosomi. Samoin tapahtuu liikkuvien elementtien sisällyttämistä genomiin ja somaattista paikallista rekombinaatiota immunoglobuliinigeenien välisessä ontogeniassa, mikä luo niiden hämmästyttävän monimuotoisuuden.
Yleisen rekombinaation virheitä voidaan pitää luonnollisina seurauksina geenien lineaarisesti pidentyneestä rakenteesta. Syntyy dilemma, josta Khesin kirjoitti: voidaan katsoa, että mitoottiset rekombinaatiot ovat erityinen mutageneesityyppi tai päinvastoin, tietyntyyppiset mutaatiot (kromosomipoikkeamat) ovat seurausta mitoottisten rekombinaatioiden "virheistä".
Jos liikkuvien elementtien liikkeet tai alueiden rekombinaatio on ohjelmoitu ontogeneesiin, on tällaisia perinnöllisiä muutoksia vaikea luokitella. Sukupuolen transformaatiota hiivassa on pitkään pidetty mutaatiotapahtumana, mutta kävi ilmi, että se tapahtuu tietyssä askosporin kehitysvaiheessa suurella todennäköisyydellä paikkaspesifisen rekombinaation seurauksena.
Genomivaihtelut vastauksena ympäristöhaasteisiin
Evoluutioteoriassa ja genetiikassa on aina keskusteltu perinnöllisten muutosten ja valinnan suunnan välisestä yhteydestä. Darwinilaisten ja postdarwinilaisten käsitysten mukaan perinnölliset muutokset tapahtuvat eri suuntiin ja vasta sitten ne poimitaan valinnalla. Erityisen selkeä ja vakuuttava oli Lederbergien 1950-luvun alussa keksimä kopiomenetelmä. Samettikankaan avulla he saivat tarkat kopiot - printit - kokeellisesta bakteerikylvyksestä petrimaljalle. Sitten yhtä levyistä käytettiin faagiresistenssin valintaan ja resistenttien bakteerien esiintymispisteiden topografiaa faagilevyllä ja kontrollissa verrattiin. Faagiresistenttien pesäkkeiden järjestely oli sama kahdessa kopiomaljassa. Sama tulos saatiin analysoitaessa positiivisia mutaatioita bakteereissa, joissa oli viallinen mikä tahansa metaboliitti.
Liikkuvan genetiikan alan löydöt ovat osoittaneet, että solu integroituneena järjestelmänä valinnan aikana voi mukauttaa genomiaan adaptiivisesti uudelleen. Se pystyy vastaamaan ympäristön haasteeseen aktiivisella geneettisellä etsinnällä, eikä odota passiivisesti mutaation sattumaa, joka mahdollistaa sen selviytymisen. Ja Lederbergin puolisoiden kokeissa soluilla ei ollut vaihtoehtoa: joko kuolema tai mukautuva mutaatio.
Tapauksissa, joissa valintatekijä ei ole tappava, ovat mahdollisia genomin asteittaiset uudelleenjärjestelyt, jotka liittyvät suoraan tai epäsuorasti valintaolosuhteisiin. Tämä tuli selväksi, kun 1970-luvun lopulla havaittiin asteittainen lisääntyminen niiden lokusten määrässä, joissa sijaitsevat geenit, jotka ovat vastustuskykyisiä selektiiviselle aineelle, joka estää solujen jakautumisen. Tiedetään, että metotreksaattia, solunjakautumisen estoainetta, käytetään laajalti lääketieteessä estämään pahanlaatuisten solujen kasvu. Tämä solumyrkky inaktivoi di(DHFR), jota säätelee tietty geeni.
Leishmania-solujen resistenssi sytostaattiselle myrkkylle (metotreksaatille) kasvoi asteittain, ja monistettujen segmenttien osuus resistenssigeenillä kasvoi vastaavasti. Ei vain valittu geeni, vaan myös sen vieressä olevat suuret DNA-alueet, joita kutsutaan amplikoneiksi. Kun Leishmanian myrkkyresistenssi lisääntyi 1000-kertaiseksi, monistetut ekstrakromosomaaliset segmentit muodostivat 10 % solun DNA:sta! Voidaan sanoa, että yhdestä pakollisesta geenistä muodostui fakultatiivisten elementtien pooli. Valinnan aikana tapahtui genomin adaptiivinen uudelleenjärjestely.
Jos valinta jatkui riittävän pitkään, osa amplikoneista siirrettiin alkuperäiseen kromosomiin, ja valinnan lopettamisen jälkeen lisääntynyt vastustuskyky säilyi.
Kun valikoiva aine poistettiin alustasta, resistenssigeenin sisältävien amplikonien määrä väheni vähitellen useissa sukupolvissa ja samalla resistenssi väheni. Näin mallinnettiin pitkäaikaisten muutosten ilmiötä, jolloin ympäristön aiheuttamat massiiviset muutokset periytyvät, mutta häviävät vähitellen useiden sukupolvien aikana.
Toistuvan valinnan aikana osa sytoplasmaan jääneistä amplikoneista varmisti nopean autonomisen replikaationsa ja vastustuskyky syntyi paljon nopeammin kuin kokeiden alussa. Toisin sanoen säilyneiden amplikonien pohjalta muodostui eräänlainen soluamplikonimuisti menneestä valinnasta.
Jos vertaamme replikoiden menetelmää ja resistenssin valinnan kulkua amplifikaatiossa, niin käy ilmi, että se oli kosketus valikoivaan tekijään, joka aiheutti genomin transformaation, jonka luonne korreloi intensiteetin ja valinnan suunta.
Keskustelua adaptiivisista mutaatioista
Vuonna 1988 Nature-lehdessä ilmestyi J. Cairnsin ja muiden kirjoittajien artikkeli valinnasta riippuvien "suunnattujen mutaatioiden" esiintymisestä E. coli -bakteerissa. Otimme bakteereita, jotka kantavat mutaatioita laktoosioperonin lacZ-geenissä, jotka eivät pystyneet hajottamaan disakkaridia laktoosia. Nämä mutantit pystyivät kuitenkin jakautumaan glukoosia sisältävällä alustalla, josta ne siirrettiin selektiiviseen laktoosialustaan yhden tai kahden päivän kasvun jälkeen. Valittuaan lac+-käänteiset, jotka odotetusti syntyivät "glukoosin" jakautumisen aikana, ei-kasvavat solut jätettiin hiilihydraattinälän olosuhteisiin. Ensin mutantit kuolivat. Mutta viikon tai useamman jälkeen havaittiin uusi kasvu johtuen lacZ-geenin reversioiden puhkeamisesta. Ikään kuin vakavassa stressissä olevat solut, ilman jakautumista (!), tekivät geneettisen haun ja muuttivat adaptiivisesti genomiaan.
B. Hallin myöhemmissä tutkimuksissa käytettiin tryptofaanin käyttögeenissä (trp) mutatoituneita bakteereja. Ne asetettiin tryptofaanittomalle alustalle ja arvioitiin palautumistiheys normaaliksi, mikä lisääntyi juuri tryptofaaninälkään. Kuitenkaan itse nälkäolosuhteet eivät olleet syynä tähän ilmiöön, koska kysteiinin nälkää sisältävässä elatusaineessa trp+:n palautumistiheys ei poikennut normaalista.
Seuraavassa koesarjassa Hall otti kaksois tryptofaanipuutteelliset mutantit, joissa oli sekä mutaatioita trpA- että trpB-geeneissä, ja asetti bakteerit uudelleen tryptofaanittomalle alustalle. Vain yksilöt, joissa reversiot tapahtuivat samanaikaisesti kahdessa tryptofaanigeenissä, voivat selviytyä. Tällaisten yksilöiden esiintymistiheys oli 100 miljoonaa kertaa odotettua suurempi yksinkertaisella todennäköisyydellä kahden geenin mutaatioiden sattuessa. Hall halusi kutsua tätä ilmiötä "adaptiivisiksi mutaatioiksi" ja osoitti myöhemmin, että niitä esiintyy myös hiivassa, ts. eukaryooteissa.
Cairnsin ja Hallin julkaisut herättivät heti kiivasta keskustelua. Sen ensimmäisen kierroksen tuloksena esiteltiin yksi mobiiligenetiikan alan johtavista tutkijoista J. Shapiro. Hän käsitteli lyhyesti kahta pääideaa. Ensinnäkin solu sisältää biokemiallisia komplekseja tai "luonnollisia geenitekniikan" järjestelmiä, jotka pystyvät muokkaamaan genomia. Näiden kompleksien toiminta, kuten kaikki muutkin solujen toiminta, voi muuttua dramaattisesti riippuen solun fysiologiasta. Toiseksi perinnöllisten muutosten esiintymistiheyttä ei aina arvioida yhdelle solulle, vaan solupopulaatiolle, jossa solut voivat vaihtaa perinnöllistä tietoa keskenään. Lisäksi solujen välinen horisontaalinen siirto virusten avulla tai DNA-segmenttien siirto tehostuu stressaavissa olosuhteissa. Shapiron mukaan nämä kaksi mekanismia selittävät ilmiön adaptiivisia mutaatioita ja palauttaa se perinteisen molekyyligenetiikan valtavirtaan. Mitkä ovat hänen mielestään keskustelun tulokset? "Löysimme sieltä geenitekniikan, jolla on vaikuttava valikoima monimutkaisia molekyylityökaluja DNA-molekyylin uudelleenjärjestelyyn." .
Viime vuosikymmeninä solutasolla on avautunut odottamaton monimutkaisuuden ja koordinaation valtakunta, joka on paremmin yhteensopiva tietokonetekniikan kuin neo-darwinilaisen modernin synteesin luomista hallinneen mekaanisen lähestymistavan kanssa. Shapiron jälkeen voidaan nimetä ainakin neljä ryhmää löytöjä, jotka ovat muuttaneet ymmärrystä solubiologisista prosesseista.
genomin järjestäytyminen. Eukaryooteissa geneettiset lokukset on järjestetty modulaarisen periaatteen mukaisesti, mikä edustaa koko genomille yhteisiä säätely- ja koodausmoduulien rakenteita. Tämä varmistaa uusien rakenteiden nopean kokoamisen ja geenikokoonpanojen säätelyn. Lokukset on organisoitu hierarkkisiin verkkoihin, joita johtaa pääkytkingeeni (kuten sukupuolen säätelyn tai silmien kehityksen tapauksessa). Lisäksi monet alisteisista geeneistä ovat integroituneet erilaisiin verkostoihin: ne toimivat eri kehitysvaiheissa ja vaikuttavat moniin fenotyypin piirteisiin.Ympäristön kutsun olosuhteissa solu toimii tarkoituksellisesti, kuten tietokone, kun sitä käynnistettäessä pääohjelmien normaali toiminta tarkistetaan askel askeleelta ja toimintahäiriön sattuessa tietokone pysähtyy. . Yleisesti ottaen käy ilmi, jo solutasolla, että epäsovinnainen ranskalainen evoluutioeläintieteilijä Paul Grasset on oikeassa: "Eläminen on reagoimista, ei uhria olemista."solun korjaavat ominaisuudet. Solut eivät suinkaan ole satunnaisten fysikaalisten ja kemiallisten vaikutusten passiivisia uhreja, koska niillä on replikaation, transkription ja translaation tasolla korjausjärjestelmä.
mobiili geneettisiä elementtejä ja luonnollinen geenitekniikka. Immuunijärjestelmän toiminta perustuu uusien immunoglobuliinimolekyylien muunnelmien jatkuvaan rakentamiseen, joka perustuu luonnollisten bioteknisten järjestelmien toimintaan (entsyymit: nukleaasit, ligaasit, käänteiskopioijat, polymeraasit jne.). Nämä samat järjestelmät käyttävät mobiilielementtejä uusien perittyjen rakenteiden luomiseen. Samaan aikaan geneettiset muutokset voivat olla massiivisia ja määrättyjä. Genomin uudelleenjärjestely on yksi tärkeimmistä biologisista prosesseista. Luonnollisia geenitekniikan järjestelmiä säätelevät palautejärjestelmät. Toistaiseksi ne ovat passiivisia, mutta avainaikoina tai stressin aikana ne aktivoituvat.
Mobiilitietojen käsittely. Ehkä yksi tärkeimmistä löydöistä solubiologiassa on se, että solu kerää ja analysoi jatkuvasti tietoa sisäisestä tilastaan ja ulkoisesta ympäristöstään ja tekee kasvua, liikettä ja erilaistumista koskevia päätöksiä. Erityisen suuntaa-antavia ovat kasvun ja kehityksen taustalla olevat solujen jakautumisen hallintamekanismit. Mitoosiprosessi on universaali korkeammissa organismeissa ja sisältää kolme peräkkäistä vaihetta: valmistautuminen jakautumiseen, kromosomien replikaatio ja solun jakautumisen loppuun saattaminen. Näiden vaiheiden geenikontrollin analyysi johti löytöyn yksittäisiä pisteitä, jossa solu tarkistaa, onko DNA-rakenteen vikojen korjaus tapahtunut edellisessä vaiheessa vai ei. Jos virheitä ei korjata, seuraava vaihe ei ala. Kun vauriota ei voida poistaa, käynnistetään geneettisesti ohjelmoitu solukuoleman eli apoptoosin järjestelmä.
Luonnollisten perinnöllisten muutosten esiintymistavat järjestelmässä ympäristö - fakultatiiviset elementit - pakolliset elementit. Fakultatiiviset elementit havaitsevat ensimmäisinä ei-mutageeniset ympäristötekijät, ja sitten syntyvät muunnelmat aiheuttavat mutaatioita. Pakolliset elementit vaikuttavat myös valinnaisten elementtien toimintaan.
Ei-kanoniset perinnölliset muutokset, jotka syntyvät sytostaattien valinnan vaikutuksesta ja johtavat geenin monistumiseen.
Hankitut ominaisuudet periytyvät
"Biologian historia ei tunne ilmaisullisempaa esimerkkiä vuosisatoja vanhasta ongelmakeskustelusta kuin keskustelu hankittujen ominaisuuksien periytymisestä tai perimättömyydestä",- nämä sanat ovat kuuluisan sytologin ja biologian historioitsijan L. Ya. Blyakherin kirjan alussa. Ehkä historiassa voidaan muistaa samanlainen tilanne yrityksillä muuttaa kemiallisia alkuaineita. Alkemistit uskoivat tähän mahdollisuuteen, mutta muuttumattomuuden postulaatti vahvistettiin kemiassa kemiallisia alkuaineita. Nyt kuitenkin sisään ydinfysiikka ja kemia, alkuaineiden muuntumisen tutkimus ja niiden evoluution analysointi on yleistä. Kuka oli oikeassa vuosisatoja vanhassa kiistassa? Voidaan sanoa, että kemiallisten molekyylivuorovaikutusten tasolla ei tapahdu alkuaineiden muutosta, mutta ydintasolla se on sääntö.
Samanlainen analogia syntyy kysymyksen ontogeneesin aikana ilmenneiden piirteiden periytymisestä. Jos äskettäin ilmenevät perinnölliset muutokset pelkistyvät vain geenien ja kromosomien mutaatioihin, voidaan kysymystä pitää suljettuna. Mutta jos lähdetään yleistetystä genomin käsitteestä, mukaan lukien ajatus dynaamisesta perinnöllisyydestä [ , ], ongelmaa on tarkistettava. Mutaatioiden lisäksi perinnöllisillä variaatioilla on muunnelmia ja epigeneettisiä muotoja, jotka eivät liity DNA-tekstin, vaan geenin tilan muutoksiin. Tällaiset vaikutukset ovat palautuvia ja periytyviä.
Mielenkiintoista on, että International Yearbook on Genetics, joka julkaistiin vuoden 1991 lopussa, alkaa O. Landmanin artikkelilla "Hankittujen piirteiden perinnöllisyys". Kirjoittaja tiivistää kauan sitten genetiikassa saadut tosiasiat osoittaen sen "hankittujen ominaisuuksien periytyminen on täysin yhteensopivaa nykyaikaisen molekyyligenetiikan käsitteen kanssa." Landman tarkastelee yksityiskohtaisesti noin kymmentä kokeellista järjestelmää, joissa hankittujen ominaisuuksien periytyminen on todettu. Siihen voi johtaa neljä erilaista mekanismia: solukalvon eli aivokuoren rakenteiden muutos, jonka T. Sonneborn on tutkinut väreissä; DNA-muunnoksia, ts. kloonisesti välittyneet muutokset paikallisen DNA:n metylaation luonteessa (tämä sisältää imprinting-ilmiön); epigeneettiset muutokset ilman DNA-muutoksia; valinnaisten osien aiheuttama menetys tai hankinta.
Landmanin artikkeli tekee meistä ikään kuin todistajia genetiikan oletusmuutosten kriittisestä ajanjaksosta, joka näytti horjumattomalta kuin kivi. Kirjoittaja rauhallisesti, ilman jännitystä ja uusia upeita faktoja, yhdistää vanhat ja uudet tiedot järjestelmäksi, antaa niille selkeän modernin tulkinnan. On mahdollista muotoilla yleinen periaate: hankittujen ominaisuuksien periytyminen on mahdollista tapauksissa, joissa tietty fenotyyppinen ominaisuus riippuu fakultatiivisten elementtien lukumäärästä tai topografiasta.
Annan kaksi opettavaa esimerkkiä Drosophilasta: ensimmäinen liittyy sigmaviruksen käyttäytymiseen, toinen - liikkuviin elementteihin, jotka ovat vastuussa naaraan hybridi-steriiliydestä ja supermutatiivisuudesta.
Sigmaviruksen ja Drosophilan genomin vuorovaikutuksen tutkimus alkoi yli 60 vuotta sitten. Ensin vuonna 1937 ranskalainen geneetikko F. Leritje havaitsi eri kärpäslinjoissa teräviä perinnöllisiä eroja herkkyyden asteen suhteen. hiilidioksidi(CO 2 ). Ominaisuus periytyi oudolla tavalla: sytoplasman kautta, mutta ei vain äidin linjan kautta, vaan joskus myös urosten kautta. Herkkyys voi tarttua myös hemolymfi-injektiolla ja erityyppisiin hedelmäkärpäsiin. Näissä tapauksissa ominaisuus ei siirtynyt vakaasti, mutta valinnan seurauksena perinnöstä tuli vakaa.
Drosophilan piirteen ei-mendeliläinen periytyminen, joka riippuu genomin fakultatiivisten elementtien populaatiosta. Merkki CO 2 -herkkyydestä johtuu rabdoviruksen sigman esiintymisestä kärpäsen sytoplasmassa. Drosophilan varhaisessa kehitysvaiheessa tapahtuneen lämpötilasokin seurauksena viruksen lisääntyminen estyy, ja aikuiset yksilöt saavat vastustuskyvyn sille.Herkkyys CO 2:lle yhdistettiin RNA:ta sisältävän luodinmuotoisen rabdoviruksen sigman vakaaseen lisääntymiseen itu- ja somaattisissa soluissa, joka on useilta ominaisuuksiltaan samanlainen kuin raivotautiviruksella nisäkkäillä. Oogonia (solut, joista munat muodostuvat meioosin ja kypsymisen aikana) stabiloidun linjan naarailla sisältävät yleensä 10-40 viruspartikkelia ja munasoluja (kypsiä munia) - 1-10 miljoonaa.Sigmavirus on tyypillinen valinnainen alkuaine. Sen genomin mutaatiot johtavat monimutkaisiin järjestelmän käyttäytymisen muotoihin. Viruksen kantajista on löydetty tapauksia, joissa Drosophila pysyy vastustuskykyisenä CO 2 :lle, mutta samalla immuuni muiden viruskantojen infektioille. Tilanne on melko verrattavissa faagi-bakteerijärjestelmän käyttäytymiseen, jonka heti huomasivat F. Jacob ja E. Volman.
Drosophilan genomin ja sen sytoplasmassa lisääntyvän viruksen välinen suhde noudattaa solunsisäisen genetiikan sääntöjä. Ontogeneesissä tapahtuvat vaikutukset voivat aiheuttaa muutosta hiukkasten lukumäärässä ja solujen välisessä topografiassa ja sen seurauksena muuttaa herkkyysastetta hiilidioksidille. Siten kohonnut lämpötila estää viruspartikkelien replikaation. Jos naaraat ja urokset pidetään 30 °C:n lämpötilassa useita päiviä gametogeneesin aikana, tällaisten kärpästen jälkeläiset ovat vapaita viruksesta ja vastustuskykyisiä CO 2 :lle. Joten hankittu aikana yksilöllistä kehitystä ominaisuus periytyy useiden sukupolvien aikana.
Sigma-viruksen tilanne ei ole yksittäinen. Ranskalaiset geneetikot tutkivat naisten hedelmättömyystekijöitä, jotka liittyvät I-tyypin liikkuvien elementtien käyttäytymiseen. Tämän ominaisuuden periytymisen määräävät monimutkaiset tuma-sytoplasmiset vuorovaikutukset. Jos aktiiviset I-elementit sijaitsevat isän kromosomeissa, ne alkavat aktivoitua R-sytoplasman taustalla, läpikäyvät useita transpositioita ja aiheuttavat sen seurauksena teräviä häiriöitä ontogeniassa herkän sytoplasman omaavien naaraiden jälkeläisissä. Tällaiset naaraat munivat, mutta osa alkioista kuolee pilkkomisen varhaisessa vaiheessa - jopa ennen blastomeerin muodostumista. Luonnollisista populaatioista eristetyt linjat eroavat I-tekijöiden vahvuudesta ja sytoplasman reaktiivisuusasteesta (tai herkkyydestä). Näitä indikaattoreita voi muuttaa ulkoinen vaikutus. Alkuperäisten vanhempien naaraiden ikä, samoin kuin kohonneen lämpötilan vaikutus varhaisen kehitysvaiheen aikana, ei vaikuta vain aikuisten naaraiden hedelmällisyyteen, vaan myös niiden jälkeläisten hedelmällisyyteen. Ympäristöolosuhteiden aiheuttamat muutokset sytoplasman reaktiivisuudessa säilyvät useiden solusukupolvien ajan. "Huomauttavinta on, että nämä muutokset sytoplasman reaktiivisuudessa ei-geneettisten tekijöiden vaikutuksesta ovat periytyviä: havaitaan "hankittujen" ominaisuuksien periytymistä",- totesi R.B. Khesin.
Periytys sytoplasman kautta: isoäideistä lastenlapsiin
1900-luvun kehitysteoriassa ja fenogenetiikassa. tärkeä paikka on embryologi P.G. Svetlovin (1892-1972) syvällisillä ja täysin alkuperäisillä tutkimuksilla. Pysähdytään hänen kehittämäänsä ontogeneesikvantisoinnin teoriaan (kriittisten jaksojen esiintyminen kehityksessä, jolloin tapahtuu morfogeneettisten prosessien määräytymistä ja samalla solujen herkkyys vaurioittaville aineille kasvaa) ja sen yhteydessä kehitettyyn ideaan. tämä, että ontogeneesin tutkimusta ei tule suorittaa hedelmöityksestä ja tsygootin muodostumisesta lähtien, eikä myöskään gametogeneesistä, mukaan lukien edellisen sukupolven naaraiden oogeneesi - proembryonaalinen ajanjakso.
Näiden postulaattien perusteella Svetlov suoritti yksinkertaisia ja selkeitä kokeita 1960-luvulla Drosophilalla ja hiirillä. Hän osoitti vakuuttavasti, että sytoplasman ominaisuuksien jatkuva ei-mendeliläinen periytyminen on mahdollista ja mutanttien ominaisuuksien vakavuuden muutokset, jotka syntyivät lyhytaikaisen ulkoisen vaikutuksen jälkeen organismin kriittisen kehitysjakson aikana, välittyvät myös sukupolvien lukumäärä.
Yhdessä koesarjassa hän vertasi mutanttiominaisuuden ilmentymisastetta kahden hiirlinjan jälkeläisissä, jotka olivat heterotsygoottisia mikroftalmian resessiivisen mutaation (verkkokalvon ja silmien pienentynyt koko syntymähetkestä lähtien) jälkeläisissä: fenotyyppi- normaalit heterotsygootit, joiden äidit olivat mutatoituneita, ja ne, joiden äidit olivat mutatoituneita, isät. Mutanttiisoäidin jälkeläiset erosivat ominaisuuden vahvemmasta ilmenemisestä. Svetlov selitti tämän oudon tosiasian sillä, että heterotsygoottisten naisten sukusolut olivat edelleen heidän mutanttiäitiensä kehossa ja vaikuttivat niihin, mikä lisäsi mutaatioita heidän lapsenlapsissaan.
Pohjimmiltaan Svetlov loi ilmiön, joka myöhemmin tunnettiin nimellä "genominen imprinting" - ero geenin ilmentymisessä riippuen siitä, tuliko se jälkeläiselle äidiltä vai isältä. Nämä teokset jäivät valitettavasti aliarvioituiksi.
Mielenkiintoista on, että jo 1980-luvun lopulla impressing, kuten tämän ilmiön tutkija K. Sapienza nokkelasti huomautti, oli "Yleisesti pidetty geneettisenä uteliaisuutena, joka vaikuttaa vain muutamaan ominaisuuteen. Minulta on toistuvasti kysytty, miksi tuhlaa aikaani niin merkityksettömään ilmiöön.. Useimmat tutkijat hyväksyivät ehdoitta yhden Mendelin pääehdotuksista - "alkeet" eli geeni ei voi muuttaa tehoaan sukupuolen mukaan, johon laajalti havaittu 3:1 jakautuminen perustuu. Mutta Sapienza huomautti aivan oikein, että analysoidessaan Mendelin halkeilua he yleensä ottavat huomioon vain piirteen olemassaolon tai puuttumisen, ja jos se on kvantitatiivista, niin rajaa. kyllä ei asetettu hyväksyttyyn kynnykseen. Jos kuitenkin ominaisuuden ilmentymisasteen paljastamiseksi paljastetaan genomisen jäljennyksen vaikutus.
Juuri tämä oli Svetlovin lähestymistapa, kun hän tutki huolellisesti, kuinka jälkeläisten ominaisuuksien vakavuus muuttuu äidin genotyypin mukaan. Emryologina hän näki perinnöllisten ja erityisten ei-perinnöllisten muutosten - fenokopioiden (simuloivien mutaatioiden) - yhteisyyden, jos sama morfogeneettinen laite, joka vastaa tietyn ominaisuuden toteuttamisesta, vaikuttaa.
Ensimmäistä kertaa eri eläinlajeissa (Drosophila ja hiiret) Svetlov osoitti mahdollisuuden periytyä mutanttigeenin ilmentymisen muuttuneen luonteen meioosin kautta. Khesin ei turhaan kutsunut näitä teoksia tiivistelmässään merkittäviksi.
Kahdeksan päivän ikäisen naarashiiren ruumiin lyhytaikainen (20 min) kuumennus aiheutti pysyviä muutoksia munasoluissa, mikä heikensi haitallisen mutaation vaikutusta lastenlapsissa! "Lämmityskokeissa havaittu silmän kehityksen paranemisen välittyminen voidaan selittää vain lämmitettyjen naaraspuolisten munasoluissa periytyvien ominaisuuksien siirtymisellä.". Svetlov liitti tämän ilmiön munan muodostumisen ja rakenteen erityispiirteisiin eläimissä, koska "Oosyytissä on ikään kuin kehys, joka heijastaa rakenteilla olevan organismin arkkitehtoniikan yleisimpiä piirteitä." Ihmisten kehityshäiriöiden ehkäisemiseksi hän perusteli tarvetta tutkia gametogeneesin kriittisiä jaksoja, jolloin herkkyys vaurioille lisääntyy. Ehkä ihmisen kehityshäiriöiden patogeneesissä sukusolujen muodostumisvaihe on jopa tärkeämpi kuin alkion muodostuminen.

P.G. Svetlovin koekaavio, joka osoittaa mutaation siirtymisen hiirten sukupolvien sarjassa - mikroftalmia. Yksittäinen 20 minuutin altistuminen kohonneelle lämpötilalle 8 päivän ikäisillä mutanttihiirillä parantaa silmän kehitystä heidän jälkeläisissään (F1 ja F2). Tämä ominaisuus periytyy vain äidin linjan kautta ja se liittyy munasolujen muutokseen.Nykyään tämän päätelmän vahvistavat viime vuosikymmenen molekyyligeneettiset tutkimukset. Drosophilassa on kolme äidingeenijärjestelmää, jotka muodostavat sytoplasman aksiaalisen ja polaarisen heterogeenisyyden ja biologisesti aktiivisten geenituotteiden jakautumisgradientit. Kauan ennen lannoituksen alkamista rakennesuunnitelman molekyylien määrittäminen (ennalta määritys) ja alkuvaiheet kehitystä. Munasolun muodostumisessa äidin organismin solujen geenituotteilla on tärkeä rooli. Eräässä mielessä tätä voidaan verrata työmehiläisten ryhmään, joka ruokkii kuningatarta pesässä.
Ihmisillä primaariset sukusolut, joista muna-sukusolut sitten syntyvät, alkavat erottua kahden kuukauden ikäisessä alkiossa. 2,5 kuukauden iässä ne siirtyvät meioosiin, mutta heti syntymän jälkeen tämä jakautuminen estyy. Se jatkuu 14–15 vuoden kuluttua murrosiän alkamisesta, jolloin munat lähtevät follikkeleista kerran kuukaudessa. Mutta toisen jakautumisen lopussa meioosi pysähtyy jälleen ja sen esto poistuu vasta kun se kohtaa siittiöiden. Siten naisten meioosi alkaa 2,5 kuukauden iässä ja päättyy vasta 20–30 vuoden kuluttua tai enemmän, heti hedelmöittymisen jälkeen.
Tsygootilla kahdesta kahdeksaan solun vaiheessa on heikentynyt genominen immuniteetti. Tutkiessamme epävakaita insertiomutaatioita Drosophilan luonnollisissa populaatioissa havaitsimme, että liikkuvan elementin aktivaatio, johon liittyy mutaatiosiirtymä, tapahtuu usein jo tsygootin ensimmäisissä jaoissa tai primääristen sukusolujen ensimmäisissä jaoissa. Tämän seurauksena yksi mutanttitapahtuma kaappaa välittömästi primaaristen sukusolujen kloonin, sukusolujen poolista tulee mosaiikki ja perinnöllisiä muutoksia jälkeläisissä tapahtuu kimppuina tai klustereina, jotka jäljittelevät perheperintöä.
Nämä kokeet ovat erittäin tärkeitä epidemiologian kannalta, kun herää kysymys tietyn virusepidemian vaikutusasteesta jälkeläisten geenipooliin. S. M. Gershenzonin ja Yu. N. Aleksandrovin 1960-luvun alussa aloittamat pioneeritutkimukset johtivat siihen johtopäätökseen, että DNA:ta ja RNA:ta sisältävät virukset ja niiden nukleiinihapot ovat voimakkaita mutageenisia tekijöitä. Päästyessään soluun ne aiheuttavat genomista stressiä, aktivoivat isännän liikkuvien elementtien järjestelmän ja aiheuttavat epävakaita insertiomutaatioita kullekin aineelle spesifisten valittujen lokusten ryhmässä.
Kuvittele nyt, että haluamme arvioida viruspandemian (esimerkiksi influenssa) vaikutusta ihmisen geneettiseen vaihteluun. Tässä tapauksessa voidaan odottaa, että taajuus erilainen kehityspoikkeamat lisääntyvät ensimmäisessä sukupolvessa vuosi tai vuosi epidemian jälkeen syntyneillä jälkeläisillä. Sukusolujen (sukusolujen) mutaatio- ja variaatiomuutosten esiintymistiheys on arvioitava lastenlapsissa.

Oogeneesikaavio kolmessa peräkkäisessä naissukupolvessa. P - isoäiti, F1 - äiti, F2 - tytär.
Yleinen johtopäätös on, että perinnöllinen vaihtelu lapsenlapsilla voi olla erittäin riippuvainen olosuhteista, joissa oogeneesi tapahtui heidän isoäideillään! Kuvittele nainen, joka vuonna 2000 oli noin 25-vuotias, ja hänestä tulee äiti kolmannella vuosituhannella. Hedelmöitetty munasolu, josta hän itse syntyi, alkoi muodostua aikana, jolloin hänen äitinsä oli vielä kahden kuukauden ikäinen alkio, ts. joskus 1950-luvun puolivälissä. Ja jos flunssa raivosi näinä vuosina, sen seurausten pitäisi tuntua sukupolvessa. Maailmanlaajuisen epidemian seurausten arvioimiseksi ihmisen geenipooliin on tarpeen verrata kolmen ryhmän eli kohortin lastenlapsia - niitä, joiden isoäidit olivat raskaana epidemian puhkeamisvuonna, niihin, joiden isoäidit tulivat raskaaksi ennen ja pandemian jälkeen (nämä ovat kaksi kontrollikohorttia). Valitettavasti tällaisia terveydensuojelun kannalta tärkeitä epidemiologisia ja geneettisiä tietoja ei ole vielä saatavilla.
Aaveista ja taistelevista hirviöistä
Kolmekymmentä vuotta on kulunut Svetlovin kokeista, jotka olivat tekniikaltaan yksinkertaisia, mutta ajatukseltaan omaperäisiä ja päätelmissään syvällisiä. 1990-luvun puolivälissä tapahtui psykologinen käännekohta: perinnöllisen vaihtelevuuden alan teosten määrä, jonka nimessä oli sana "epigeneettinen", kasvoi jyrkästi.
Erilaiset epimutaatiot (geenitoiminnan luonteen perinnölliset vaihtelut, jotka eivät liity DNA-tekstin muutoksiin ja ovat massiivisia, suunnattuja ja palautuvia) ovat siirtyneet marginaalista aktiivisesti tutkituksi ilmiöksi. Tuli ilmeiseksi, että elävillä järjestelmillä on toiminnallinen "muisti", joka on jatkuvassa yhteydessä ympäristöön ja käyttää luonnollisen alkiogeenitekniikan keinoja nopeaan periytyvään siirtymiseen toimintatavasta toiseen. Elävät järjestelmät eivät ole passiivisia uhreja luonnonvalinta, ja kaikki evolutionaariset elämänmuodot eivät ole ollenkaan "blot for a short escheat day", kuten Mandelstam kirjoitti kuuluisassa mestariteoksessaan Lamarck.
Kävi ilmi, että epimutaatioita löytyy hyvin usein tavallisista "klassisista geeneistä", sinun tarvitsee vain valita sopiva kokeellinen järjestelmä. Vuonna 1906, viisi vuotta ennen kuin Morgan aloitti työskentelyn Drosophilan kanssa, ranskalainen evoluutiobiologi L. Keno löysi Mendelin keltaisen kehon mutaation hiiristä. Hänellä oli hämmästyttävä ominaisuus - dominanssi suhteessa normaaliin väriin (harmaa-ruskea) ja kuolleisuus homotsygootissa. Kun heterotsygoottiset keltaiset hiiret risteytettiin keskenään, homotsygoottien kuoleman vuoksi normaaleja hiiriä ilmestyi jälkeläisiin suhteessa 3:1, vaan 2:1. Myöhemmin kävi ilmi, että monet hallitsevat mutaatiot eri organismeissa käyttäytyvät tällä tavalla.
Kävi ilmi, että "keltaisen kehon" geenin yhden alleelin transkriptioalueelle tuotiin liikkuva elementti, joka muistutti rakenteeltaan ja ominaisuuksiltaan retrovirusta. Tämän lisäyksen seurauksena geeni alkoi totella tunkeilijansa välimerkkejä ja aktivoitui arvaamattomasti "väärään aikaan ja väärässä paikassa." Mutantit, joilla on insertioita, kehittävät useita vikoja (keltainen turkki, lihavuus, diabetes jne.), ja niiden käyttäytyminen muuttuu epävakaaksi. Tarpeeton inserttiaktiivisuus sammuu vaihtelevassa määrin eri kudoksissa johtuen DNA-emästen palautuvasta modifikaatiosta tai metylaatiosta. Fenotyyppitasolla hallitsevan alleelin ilmenemismuoto vaihtelee suuresti ja on luonteeltaan mosaiikkinen. Australialaiset geneetikot havaitsivat, että homogeenisesta linjasta valituilla keltaisilla naarailla oli enemmän keltaisia hiiriä jälkeläisissään, ja isän - mutaation kantajan - fenotyyppi ei vaikuttanut jälkeläisten värinmuutokseen. Naaraat osoittautuivat inertiaalisemmiksi, ja ne DNA-modifikaatiofenotyypin eli jälkien mukaan valikoituina säilyivät paremmin oogeneesissä. Muut geneetikot ovat myös löytäneet puhtaasti äidin vaikutuksen, samanlaisen kuin Svetlovin kokeissa. Raskaana olevien naisten ruokavaliosta riippuen "keltaisen kehon" mutaation vakavuus muuttui tietyllä tavalla heterotsygoottien genotyypissä. Tämä muuttunut tila on epävakaa, mutta periytyy jälkeläisille. Ominaisuuden ilmenemisaste korreloi insertissä olevien DNA-emästen metylaatioasteen kanssa.
Viitaten näihin ja muihin vastaaviin kokeisiin "Science"-lehden tieteellinen arvioija kutsui artikkeliaan "Oliko Lamarck vielä vähän oikeassa?" Tämä taktiikka on ymmärrettävää. Ensinnäkin varovaisuus on perusteltua tarkistettaessa sitä, mitä on pidetty vakiintuneena vuosikymmeniä. Toiseksi hankittujen ominaisuuksien periytyminen ei liity vain Lamarckin nimeen, vaan myös Lysenkon haamuon (muistiinpanon kirjoittaja mainitsee jälkimmäisen). Itse asiassa, vapaaehtoisesti tai tahattomasti, "Michurin-biologian" varjo tulee esiin, kun keskustellaan hankittujen ominaisuuksien periytymisongelmasta. Eikä vain Venäjällä, missä muisti Lysenkon hallitsemiseen liittyvästä biologian tragediosta on edelleen elossa.
Nykyään monet yleisesti hyväksytyt klassisen genetiikan säännökset, jotka Lysenko hylkäsi, on tahattomasti, hänestä huolimatta, tullut melkein absoluuttiseksi totuudeksi. Ja kuitenkin, jos yksi tai toinen vakava tutkija havaitsi jotain Lysenkon näkemysten kanssa ulkoisesti sopusoinnussa, hän pelkäsi tuoda sen julkisuuteen peläten tiedeyhteisön syrjäytymistä. Ja vaikka teos julkaistiin, siihen liittyi monia varauksia ja se pysyi tieteen reuna-alueella.
Tutustuttuani 60-luvulla A. A. Lyubishchevin (Svetlovin lähimmän ystävän) artikkeleihin, yritin ymmärtää, miksi hänen artikkelinsa ja kirjeensä kerättiin kirjaan, koska hän oli yksi aktiivisimmista lysenkolaisuuden omakustanteisista kriitikoista vuosina 1953–1965. "Tieteen puolustamiseksi" (L., 1990) - ei kuitenkaan pitänyt hankittujen ominaisuuksien periytymistä lopullisesti ratkaistuna. Tämä yleisesti tunnustettu evoluutiobiologian asiantuntija viittasi perinnöllisyysteorian epätäydellisyyteen, perinnöllisen ja muunnelman vaihtelun samankaltaisuuteen. Nyt tiedämme, kuinka vaikeaa on monissa tapauksissa vetää raja niiden välille. Lyubishchev mainitsi tosiasiat fenotyypin massoista, nopeista ja järjestetyistä muutoksista evoluutiossa, jotka ovat selvästi käsittämättömiä Morgan-mutaatioiden ja darwinilaisen valinnan näkökulmasta. Lysenkon monopolia vastaan korotettuaan Lyubishchev puhui puolustaessaan tiedettä sellaisenaan, siihen vakiinnuttanutta Arakcheev-hallintoa vastaan. Itse tieteen alalla hän noudatti muinaista periaatetta: "Platon on ystäväni, mutta totuus on kalliimpi".
9. McClintock b.// Tiede. 1984. V.226. P.792-801.
10. Cairns J.// Luonto. 1988.V.27. P.1-6.
11. Halli D.// Genetiikka. 1990. V.126. P.5-16
12. Shapiro J.// Tiede. 1995. V.268. P.373-374.
12. Blyakher L. Ya. Hankittujen ominaisuuksien periytymisongelma. M., 1971.
13. Landman O.// Ann. Rev. Genet. 1991. V.25. P.1-20.
14. Sokolova K.B. Fenogenetiikan kehitys 1900-luvun ensimmäisellä puoliskolla. M., 1998.
15. Sapienza K.// Tieteen maailmassa. 1990. ?12. s. 14-20.
16. Svetlov P.G.// Genetiikka. 1966.?5. S.66-82.
17. Korochkin L.I. Johdatus kehitysgenetiikkaan. M., 1999.
) löytyy hedelmäkärpäsen genomista ( Drosophila ananassae) täydellinen kopio loisen bakteerigenomista Wolbachia.
Wolbachia-bakteeri elää isäntäsolujen sytoplasmassa ja tunnetaan siitä, että se on oppinut säätelemään hienosti isäntiensä lisääntymistä, kehitystä ja jopa evoluutiota. Siksi sitä kutsutaan usein "mikrobimanipulaattoriksi" tai "kärpästen herraksi" (koska se elää hyönteissoluissa).
Tutkimus alkoi, kun JCVI:n Julie Dunning-Hotopp havaitsi, kuinka tietyt Wolbachia-geenit "toimivat yhteistyössä" Drosophila-geenien kanssa ikään kuin ne olisivat osa samaa genomia.
Michael Clark - Rochesterin yliopiston tutkija - asetti siirtokunnan Drosophila ananassae laboratoriossa selvittääkseen salaisuuden Warrenin kanssa.
Wolbachia-geeni Drosophilan genomissa (kuvittanut Rochesterin yliopisto).
"Useita kuukausia luulin, että olin jossain väärässä", Clarke sanoo, "oletin jopa, että antibioottiresistenssi oli kehittynyt, koska löysin jokaisen Wolbachia-geenin uudestaan ja uudestaan. Kun vihdoin otin ne nenäliinat, jotka olin jättänyt rauhaan muutama kuukausi sitten, en löytänyt itse Wolbachiaa.
Nyt Warren ja Clarke yrittävät selvittää, mitä hyötyä niin suuren DNA-palan lisäämisestä Drosophilalle on - kenties "vieraat" geenit tarjoavat isännälle uusia mahdollisuuksia.

Ja niin Wolbachia-geenit siirtyvät isännän DNA:han (kuva Nicolle Rager Fuller, National Science).
Tutkimuksen tulokset julkaistiin Science-lehdessä julkaistussa artikkelissa. Siinä kirjoittajat ehdottavat, että horisontaalista geeninsiirtoa (geenien siirtoa toisiinsa liittyvien lajien välillä) tapahtuu maailmassamme bakteerien ja monisoluisten organismien välillä paljon useammin kuin aiemmin on ajateltu.
Wolbachian isäntiensä kanssa suorittamien manipulaatioiden molekyyligeneettisten mekanismien purkaminen antaa ihmiselle voimakkaita uusia keinoja vaikuttaa eläviin organismeihin ja luontoon kokonaisuudessaan.
Kaikki hyönteiset eivät kuitenkaan ole herkkiä huono vaikutus wolbachia. Esimerkiksi Samoan saarten perhoset ovat "oppineet" suojelemaan uroksiaan. Mietin, oppivatko malariahyttyset, jotka he haluavat tartuttaa tällä bakteerilla, taistelemaan sitä vastaan?
DNA:n rakenteen löytämisen 50-vuotispäivää
A.V. Zelenin
KASVIGENOME
A. V. Zelenin
Zelenin Aleksanteri Vladimirovitš- d.b.n.,
molekyylibiologian instituutin laboratorion johtaja. V.A. Engelhardt RAS.
"Human Genome" -ohjelman vaikuttavat saavutukset sekä ns. ekstrapienten (virukset), pienten (bakteerit, hiiva) ja keskikokoiset (suolimato, Drosophila) genomien salaustyön onnistuminen mahdollistivat siirtyä laajamittaiseen suurten ja erittäin suurien kasvien genomien tutkimukseen. Taloudellisesti tärkeimpien kasvien genomien yksityiskohtaisen tutkimuksen kiireellistä tarvetta korostettiin Yhdysvalloissa vuonna 1997 pidetyssä kasvigenomiikkaa käsittelevässä kokouksessa [ , ]. Siitä kuluneiden vuosien aikana tällä alalla on saavutettu kiistattomia menestyksiä. Vuonna 2000 ilmestyi julkaisu pienen sinapin - Arabidopsiksen - genomin täydellisestä sekvensoinnista (koko ydin-DNA:n lineaarisen nukleotidisekvenssin määritys), vuonna 2001 - riisin genomin alustavasta (luonnos) sekvensoinnista. Suurten ja supersuurien kasvigenomien (maissi, ruis, vehnä) sekvensointitöitä raportoitiin toistuvasti, mutta nämä raportit eivät sisältäneet erityistä tietoa ja olivat pikemminkin aiejulistuksia.
Kasvien genomien dekoodauksen oletetaan avaavan laajat mahdollisuudet tieteelle ja käytännölle. Ensinnäkin uusien geenien tunnistaminen ja niiden geneettisen säätelyn ketju lisäävät merkittävästi kasvien tuottavuutta bioteknisten lähestymistapojen avulla. Geenien löytäminen, eristäminen, lisääntyminen (kloonaus) ja sekvensointi, jotka vastaavat kasviorganismin sellaisista tärkeistä toiminnoista kuin lisääntyminen ja tuottavuus, vaihteluprosessit, vastustuskyky haitallisille ympäristötekijöille sekä kromosomien homologinen pariutuminen, syntyi liittyy uusia mahdollisuuksia jalostusprosessin parantamiseen. Lopuksi eristettyjä ja kloonattuja geenejä voidaan käyttää siirtogeenisten kasvien saamiseksi, joilla on täysin uusia ominaisuuksia, ja analysoida geeniaktiivisuuden säätelymekanismeja.
Kasvigenomien tutkimisen tärkeyttä korostaa myös se, että toistaiseksi lokalisoitujen, kloonattujen ja sekvensoitujen kasvigeenien määrä on pieni ja vaihtelee eri arvioiden mukaan 800 ja 1200 välillä. Tämä on 10-15 kertaa vähemmän kuin vuonna 2010. esimerkiksi ihmisissä.
Yhdysvallat on edelleen kiistaton johtaja laajamittaisessa kasvigenomitutkimuksessa, vaikka riisin genomin intensiivisiä tutkimuksia tehdään Japanissa ja viime vuosina Kiinassa. Arabidopsiksen genomin tulkitsemiseen osallistuivat yhdysvaltalaisten laboratorioiden lisäksi aktiivisesti eurooppalaiset tutkimusryhmät. Yhdysvaltojen näennäinen johtajuus aiheuttaa vakavaa huolta eurooppalaisissa tutkijoissa, minkä he ilmaisivat selvästi kokouksessa merkittävällä otsikolla "Prospects for Genomics in the Post-Genomic Era", joka pidettiin vuoden 2000 lopulla Ranskassa. Amerikkalaisen tieteen edistyminen maatalouskasvien genomien tutkimisessa ja siirtogeenisten kasvimuotojen luomisessa eurooppalaisten tutkijoiden mukaan uhkaa, että ei liian kaukaisessa tulevaisuudessa (kahdesta viiteen vuosikymmeneen), jolloin väestönkasvu asettaa ihmiskunnan yleisen tilanteen eteen. elintarvikekriisi, Euroopan talous ja tiede tulevat riippuvaisiksi amerikkalaisesta teknologiasta. Tältä osin ilmoitettiin ranskalais-saksalaisen tieteellisen ohjelman perustamisesta kasvien genomien tutkimukseen ("Plantgene") ja siihen tehtiin merkittäviä investointeja.
Ilmeisesti kasvien genomiikan ongelmien tulisi kiinnittää venäläisten tutkijoiden ja tieteen järjestäjien sekä hallintoelinten tarkkaavainen huomio, koska kyse ei ole vain tieteellisestä arvovallasta, vaan myös maan kansallisesta turvallisuudesta. Vuosikymmenen tai kahden kuluttua ruoasta tulee tärkein strateginen resurssi.
KASVIGENOMIEN TUTKIMUKSEN VAIKKEUDET
Kasvien genomien tutkiminen on paljon vaikeampi tehtävä kuin ihmisten ja muiden eläinten genomin tutkiminen. Tämä johtuu seuraavista olosuhteista:
valtavat genomikoot, jotka saavuttavat kymmeniä ja jopa satoja miljardeja emäspareja (bp) yksittäisten kasvilajien kohdalla: tärkeimpien taloudellisesti tärkeiden kasvien (riisi, pellava ja puuvilla) genomit ovat joko kooltaan lähellä ihmisen genomia tai ylittää sen monta kertaa (taulukko);GENOMIEN KROMOSOMITUTKIMUKSETKromosomien lukumäärän voimakkaat vaihtelut eri kasveissa - kahdesta joissakin lajeissa useisiin satoihin toisissa, eikä genomin koon ja kromosomien lukumäärän välillä ole mahdollista tunnistaa tiukkaa korrelaatiota;
Lukuisat polyploidimuodot (jotka sisältävät enemmän kuin kaksi genomia solua kohden) muodostavat samanlaisia, mutta eivät identtisiä genomeja (alpolyploidia);
Kasvien genomien äärimmäinen rikastuminen (jopa 99 %) "merkittämättömästä" (ei-koodaavasta, eli geenejä sisältämättömästä) DNA:sta, mikä tekee sekvensoitujen fragmenttien erittäin vaikeaksi liittyä (järjestyä oikeaan järjestykseen) yhteiseksi suurikokoinen DNA-alue (jatkuvuus);
Epätäydellinen (verrattuna Drosophilan, ihmisen ja hiiren genomiin) morfologinen, geneettinen ja fyysinen kromosomien kartoitus;
Käytännön mahdottomuus eristää yksittäisiä kromosomeja puhtaassa muodossa menetelmillä, joita tavallisesti käytetään tähän tarkoitukseen ihmisten ja eläinten kromosomeille (lajittelu virrassa ja soluhybridien käyttö);
Yksittäisten geenien kromosomikartoituksen (paikan määrittäminen kromosomissa) vaikeus hybridisaatiolla paikan päällä, johtuen sekä "merkittämättömän" DNA:n korkeasta pitoisuudesta kasvin genomissa ja kasvien kromosomien rakenteellisen järjestyksen erityispiirteistä;
Kasvien evoluutionaalinen etäisyys eläimistä, mikä vaikeuttaa vakavasti ihmisten ja muiden eläinten genomien sekvensoimalla saadun tiedon käyttöä kasvien genomien tutkimiseen;
Useimpien kasvien pitkä lisääntymisprosessi, mikä hidastaa merkittävästi niiden geneettistä analyysiä.
Yleensä genomien ja erityisesti kasvien kromosomaalisilla (sytogeneettisillä) tutkimuksilla on pitkä historia. Termillä "genomi" ehdotettiin haploidista (yksittäistä) kromosomien sarjaa niiden sisältämien geenien kanssa 1900-luvun ensimmäisellä neljänneksellä, toisin sanoen kauan ennen kuin DNA:n rooli geneettisen tiedon kantajana vakiintui. .
Uuden, aiemmin geneettisesti tutkimattoman monisoluisen organismin genomin kuvaus alkaa yleensä sen kromosomien täydellisen sarjan (karyotyypin) tutkimisesta ja kuvauksesta. Tämä koskee tietysti myös kasveja, joista suurta määrää ei ole edes aloitettu tutkimaan.
Jo kromosomitutkimusten kynnyksellä verrattiin sukulaiskasvilajien genomeja interspesifisten hybridien meioottisen konjugaation (homologisten kromosomien yhdistelmän) analyysin perusteella. Viimeisten 100 vuoden aikana kromosomianalyysin mahdollisuudet ovat laajentuneet dramaattisesti. Nyt kasvien genomien karakterisointiin käytetään kehittyneempiä tekniikoita: niin sanotun differentiaalivärjäyksen eri variantteja, jotka mahdollistavat yksittäisten kromosomien tunnistamisen morfologisten ominaisuuksien perusteella; hybridisaatio paikan päällä spesifisten geenien paikallistamisen mahdollistaminen kromosomeihin; soluproteiinien biokemialliset tutkimukset (elektroforeesi ja immunokemia) ja lopuksi joukko menetelmiä, jotka perustuvat kromosomaalisen DNA:n analyysiin sen sekvensointiin asti.

Riisi. 1. Viljan karyotyypit a - ruis (14 kromosomia), b - durumvehnä (28 kromosomia), c - pehmeä vehnä (42 kromosomia), d - ohra (14 kromosomia)Viljojen, pääasiassa vehnän ja rukiin, karyotyyppejä on tutkittu useiden vuosien ajan. Mielenkiintoista on, että näiden kasvien eri lajeissa kromosomien lukumäärä on erilainen, mutta aina seitsemän kerrannainen. Yksittäiset viljatyypit voidaan tunnistaa luotettavasti niiden karyotyypin perusteella. Esimerkiksi rukiin genomi koostuu seitsemästä parista suuria kromosomeja, joiden päissä on voimakkaan värisiä heterokromaattisia lohkoja, joita usein kutsutaan segmenteiksi tai vyöhykkeiksi (kuva 1a). Vehnägenomeissa on jo 14 ja 21 kromosomiparia (kuvat 1, b, c), ja heterokromaattisten lohkojen jakautuminen niissä ei ole sama kuin rukiin kromosomeissa. Yksittäiset vehnän genomit, merkinnällä A, B ja D, eroavat myös toisistaan. Kromosomien lukumäärän kasvu 14:stä 21:een johtaa vehnän ominaisuuksien jyrkkään muutokseen, mikä näkyy niiden nimissä: durum, tai pastaa, vehnää ja pehmeää, tai leipää, vehnää . Gluteeniproteiinien geenejä sisältävä D-geeni, joka antaa taikinalle ns. itävyyden, vastaa siitä, että pehmeä vehnä saa korkeat leivontaominaisuudet. Juuri tähän genomiin kiinnitetään erityistä huomiota leipävehnän valinnan parantamisessa. Toista 14-kromosomista viljaa, ohraa (kuva 1, d), ei yleensä käytetä leivän valmistukseen, mutta se on pääraaka-aine yleisten tuotteiden kuten oluen ja viskin valmistuksessa.
Joidenkin tärkeimpien maatalouslajien, kuten vehnän luonnonvaraisten sukulaisten - Aegilopsin, laadun parantamiseen käytettyjen luonnonvaraisten kasvien kromosomeja tutkitaan intensiivisesti. Uusia kasvimuotoja luodaan risteyttämällä (kuva 2) ja valinnalla. Viime vuosina tutkimusmenetelmien merkittävä parannus on mahdollistanut sellaisten kasvigenomien tutkimuksen aloittamisen, joiden karyotyyppien ominaisuudet (lähinnä kromosomien pieni koko) tekivät niistä aiemmin saavuttamattomissa kromosomianalyysissä. Joten vasta äskettäin kaikki puuvillan, kamomillan ja pellavan kromosomit tunnistettiin ensimmäistä kertaa.

Riisi. 2. Vehnän karyotyypit ja vehnän hybridi Aegilopsin kanssa
a - heksaploidinen pehmeä vehnä ( Triticum astivum), joka koostuu A-, B- ja O-genomeista; b - tetraploidinen vehnä ( Triticum timopheevi), joka koostuu A- ja G-genomeista. sisältää geenejä, jotka ovat vastustuskykyisiä useimmille vehnäsairauksille; c - hybridit Triticum astivum X Triticum timopheevi vastustuskykyinen härmäsientä ja ruostetta vastaan, osan kromosomista uusiutuminen on selvästi näkyvissäDNA:N ENSISIJAINEN RAKENNE
Molekyyligenetiikan kehittymisen myötä genomin käsite on laajentunut. Nyt tätä termiä tulkitaan sekä klassisessa kromosomaalisessa että nykyaikaisessa molekyylisessä mielessä: yksittäisen viruksen, solun ja organismin koko geneettinen materiaali. Luonnollisesti useiden mikro-organismien ja ihmisten genomien täydellisen primäärirakenteen (kuten nukleiinihappoemästen täydellistä lineaarista sekvenssiä usein kutsutaan) tutkimuksen jälkeen nousi esiin kysymys kasvien genomin sekvensoinnista.
Monista kasviorganismeista kaksi valittiin tutkimukseen - Arabidopsis, joka edustaa kaksisirkkaisten luokkaa (genomin koko 125 miljoonaa bp), ja riisi yksisirkkaisten luokasta (420-470 miljoonaa bp). Nämä genomit ovat pieniä verrattuna muihin kasvigenomeihin ja sisältävät suhteellisen vähän toistuvia DNA-segmenttejä. Tällaiset ominaisuudet antoivat toivoa, että valitut genomit olisivat käytettävissä niiden primäärirakenteen suhteellisen nopeaa määritystä varten.

Riisi. 3. Arabidopsis - pieni sinappi - pieni kasvi ristikukkaisten heimosta ( Brassicaceae). Lehden yhden sivun pinta-alalla voit kasvattaa jopa tuhatta yksittäistä Arabidopsis-organismia.Syynä Arabidopsiksen valintaan ei ollut vain sen genomin pieni koko, vaan myös organismin pieni koko, mikä tekee siitä helppoa kasvattaa laboratoriossa (kuva 3). Otimme huomioon sen lyhyen lisääntymissyklin, jonka ansiosta on mahdollista tehdä nopeasti risteyttämis- ja valintakokeita, yksityiskohtaisesti tutkittu genetiikka, helppokäyttöisyys muuttuvissa kasvuolosuhteissa (maaperän suolakoostumuksen muuttaminen, erilaisten ravinteiden lisääminen jne. .) ja erilaisten mutageenisten tekijöiden ja patogeenien (virukset, bakteerit, sienet) vaikutusten testaaminen kasveissa. Arabidopsiksella ei ole taloudellista arvoa, joten sen genomia kutsuttiin hiiren genomin ohella viitteeksi tai, vähemmän tarkasti, malliksi.*
* Termin "malligenomi" esiintyminen venäläisessä kirjallisuudessa on seurausta englanninkielisen lauseen malli genomi virheellisestä käännöksestä. Sana "malli" ei tarkoita vain adjektiivia "malli", vaan myös substantiivia "näyte", "standardi", "malli". Olisi oikeampaa puhua näytegenomista tai referenssigenomista.Intensiivinen työ Arabidopsis-genomin sekvensointiin aloitti vuonna 1996 kansainvälinen konsortio, johon kuului tieteellisiä instituutioita ja tutkimusryhmiä Yhdysvalloista, Japanista, Belgiasta, Italiasta, Isosta-Britanniasta ja Saksasta. Joulukuussa 2000 saatiin laajaa tietoa Arabidopsis-genomin primäärirakenteen määrittämisestä. Sekvensoinnissa käytettiin klassista tai hierarkkista tekniikkaa: ensin tutkittiin yksittäisiä pieniä genomin osia, joista koostui suurempia osia (contigeja) ja viimeisessä vaiheessa yksittäisten kromosomien rakennetta. Arabidopsis-genomin ydin-DNA on jakautunut viiteen kromosomiin. Vuonna 1999 julkaistiin kahden kromosomin sekvensoinnin tulokset, ja lehdistössä ilmestynyt tieto kolmen jäljellä olevan kromosomin primäärirakenteesta saattoi koko genomin sekvensoinnin päätökseen.
125 miljoonasta emäsparista on määritetty 119 miljoonan primäärirakenne, mikä on 92 % koko genomista. Vain 8 % Arabidopsis-genomista, joka sisälsi suuria lohkoja toistuvia DNA-segmenttejä, osoittautui mahdottomaksi tutkia. Eukaryoottisen genomin sekvensoinnin täydellisyyden ja perusteellisuuden suhteen Arabidopsis on edelleen kolmen parhaan mestarin joukossa yksisoluisen hiivaorganismin kanssa. Saccharomyces cerevisiae ja monisoluinen organismi Caenorhabditis eleganssia(katso taulukko).
Arabidopsiksen genomista on löydetty noin 15 000 yksittäistä proteiinia koodaavaa geeniä. Näistä noin 12 000 sisältyy kahtena kopiona per haploidi (yksittäinen) genomi, joten geenien kokonaismäärä on 27 000. Arabidopsiksen geenien määrä ei juuri poikkea geenien määrästä eliöissä, kuten ihmisissä ja hiirissä, mutta genomin koko on 25-30 kertaa pienempi. Tämä seikka liittyy tärkeisiin piirteisiin yksittäisten Arabidopsis-geenien rakenteessa ja sen genomin yleisessä rakenteessa.
Arabidopsis-geenit ovat kompakteja, ja ne sisältävät vain muutaman eksonin (proteiinia koodaavia alueita), jotka on erotettu lyhyillä (noin 250 bp) ei-koodaavilla DNA-segmenteillä (introneilla). Yksittäisten geenien väliset välit ovat keskimäärin 4600 emäsparia. Vertailun vuoksi huomautamme, että ihmisen geenit sisältävät useita kymmeniä ja jopa satoja eksoneja ja introneita, ja geenien välisten alueiden koko on 10 tuhatta emäsparia tai enemmän. Oletetaan, että pienen kompaktin genomin läsnäolo vaikutti Arabidopsiksen evolutionaariseen stabiilisuuteen, koska sen DNA:sta tuli vähemmässä määrin kohde useille vahingollisille aineille, erityisesti viruksen kaltaisten toistuvien DNA-fragmenttien (transposonien) viemiselle. genomiin.
Muiden Arabidopsis-genomin molekyyliominaisuuksien joukossa on huomattava, että eksonit ovat rikastettuja guaniinilla ja sytosiinilla (44 % eksoneissa ja 32 % introneissa) verrattuna eläingeeneihin, sekä kaksinkertaisesti toistuvien (kaksoistettujen) geenien läsnäolo. Oletetaan, että tällainen kaksinkertaistuminen tapahtui neljän samanaikaisen tapahtuman seurauksena, jotka koostuvat Arabidopsis-geenien osan kaksinkertaistumisesta (toistosta) tai sukulaisten genomien fuusiosta. Nämä tapahtumat, jotka tapahtuivat 100-200 miljoonaa vuotta sitten, ovat ilmentymä yleisestä polyploidisaatiotrendistä (organismin genomien lukumäärän moninkertainen lisääntyminen), joka on tyypillistä kasvigenomeille. Jotkut tosiasiat osoittavat kuitenkin, että Arabidopsiksen kaksoisgeenit eivät ole identtisiä ja toimivat eri tavalla, mikä voi liittyä mutaatioihin niiden säätelyalueilla.
Riisistä on tullut toinen täydellisen DNA-sekvensoinnin kohde. Tämän kasvin genomi on myös pieni (12 kromosomia, yhteensä 420-470 miljoonaa bp), vain 3,5 kertaa suurempi kuin Arabidopsiksen. Toisin kuin Arabidopsis, riisillä on kuitenkin suuri taloudellinen merkitys, sillä se on ravinnon perusta yli puolelle ihmiskunnasta, joten miljardien kuluttajien lisäksi myös monimiljoonainen armeija osallistuu aktiivisesti sen erittäin työläiseen prosessiin. viljely on elintärkeästi kiinnostunut sen ominaisuuksien parantamisesta.
Jotkut tutkijat alkoivat tutkia riisin genomia jo 1980-luvulla, mutta nämä tutkimukset saavuttivat vakavan mittakaavan vasta 1990-luvulla. Vuonna 1991 Japanissa luotiin ohjelma riisin genomin rakenteen tulkitsemiseksi, ja se kokosi yhteen monien tutkimusryhmien ponnistelut. Tämän ohjelman pohjalta järjestettiin vuonna 1997 International Rice Genome Project. Sen osallistujat päättivät keskittää ponnistelunsa riisin yhden alalajin sekvensointiin ( Oriza sativajaponica), jonka tutkimuksessa oli jo tuolloin saavutettu merkittävää edistystä. Vakava ärsyke ja kuvaannollisesti sanottuna johtotähti sellaiselle työlle oli "Human Genome" -ohjelma.
Tämän ohjelman puitteissa testattiin genomin "kromosomaalisen" hierarkkisen jaon strategiaa, jota kansainvälisen konsortion osallistujat käyttivät riisin genomin tulkinnassa. Kuitenkin, jos ihmisen genomin tutkimuksessa eristettiin yksittäisten kromosomien fraktioita eri menetelmillä, niin yksittäisille riisikromosomeille ja niiden yksittäisille alueille spesifinen materiaali saatiin lasermikrodissektiolla (mikroskooppisten esineiden leikkaaminen). Mikroskoopin objektilasilla, jossa on riisin kromosomit sijaitsevat, lasersäteen vaikutuksesta kaikki palaa, paitsi kromosomi tai sen analyysiin varatut osat. Jäljelle jäävää materiaalia käytetään kloonaukseen ja sekvensointiin.
Lukuisia raportteja on julkaistu riisin genomin yksittäisten fragmenttien sekvensoinnin tuloksista, jotka on suoritettu suurella tarkkuudella ja yksityiskohtaisesti, mikä on ominaista hierarkkiselle teknologialle. Riisigenomin täydellisen primäärirakenteen uskottiin valmistuvan vuoden 2003 loppuun mennessä – vuoden 2004 puoliväliin mennessä, ja tuloksia sekä Arabidopsis-genomin primäärirakennetta koskevien tietojen kanssa käytetään laajasti vertailussa. muiden kasvien genomiikka.
Kuitenkin vuoden 2002 alussa kaksi tutkimusryhmää - toinen Kiinasta, toinen Sveitsistä ja Yhdysvalloista - julkaisi tulokset täydellisestä (likimääräisestä) riisin genomin sekvensointiluonnoksesta, joka suoritettiin käyttämällä kokonaiskloonaustekniikkaa. Toisin kuin vaiheittaisessa (hierarkkisessa) tutkimuksessa, kokonaislähestymistapa perustuu koko genomisen DNA:n samanaikaiseen kloonaukseen johonkin virus- tai bakteerivektoreista ja merkittävän (valtava määrä keskikokoisille ja suurille genomeille) yksittäisiä klooneja, jotka sisältävät erilaisia DNA-segmentit. Näiden sekvensoitujen osien analyysin ja DNA:n identtisten terminaalisten osien päällekkäisyyden perusteella muodostuu contig - DNA-sekvenssien ketju, jotka on liitetty yhteen. Yleinen (kokonais) jatkumo on koko genomin tai ainakin yksittäisen kromosomin ensisijainen rakenne.
Tällaisessa kaavamaisessa esityksessä kokonaiskloonauksen strategia näyttää yksinkertaiselta. Itse asiassa se kohtaa vakavia vaikeuksia, jotka liittyvät tarpeeseen hankkia valtava määrä klooneja (on yleisesti hyväksytty, että tutkittava genomi tai sen alue on päällekkäinen kloonien kanssa vähintään 10 kertaa), valtava määrä sekvensointia ja erittäin monimutkainen. kloonien telakointityö, joka edellyttää bioinformatiikan asiantuntijoiden osallistumista. Vakava este täydelliselle kloonaukselle ovat erilaiset toistuvat DNA-segmentit, joiden määrä, kuten jo mainittiin, kasvaa jyrkästi genomin koon kasvaessa. Siksi kokonaissekvensoinnin strategiaa käytetään pääasiassa virusten ja mikro-organismien genomien tutkimuksessa, vaikka sitä onkin menestyksekkäästi käytetty monisoluisen organismin, Drosophilan, genomin tutkimiseen.
Tämän genomin kokonaissekvensoinnin tulokset "asetettiin" valtavalle joukolle tietoa sen kromosomi-, geeni- ja molekyylirakenteesta, jotka saatiin lähes 100 vuoden Drosophilan tutkimusjakson aikana. Ja silti, sekvensointiasteen suhteen, Drosophilan genomi (66% genomin kokonaiskoosta) on huomattavasti huonompi kuin Arabidopsis-genomi (92%) huolimatta niiden melko läheisestä koosta - 180 miljoonaa ja 125 miljoonaa emäsparia, vastaavasti. . Siksi on äskettäin ehdotettu nimeämistä sekateknologialle, jota käytettiin Drosophilan genomin sekvensointiin.
Riisin genomin sekvensoimiseksi edellä mainitut tutkimusryhmät ottivat sen kaksi alalajia, laajimmin viljeltyjä Aasian maissa, - Oriza saliva L. ssp indicaj Ja Oriza saliva L. sspjaponica. Heidän tutkimustuloksensa ovat monessa suhteessa samat, mutta poikkeavat monilta osin. Siten molempien ryhmien edustajat ilmoittivat saavuttaneensa noin 92-93 % genomin päällekkäisyydestä jatkuvien kanssa. On osoitettu, että noin 42 % riisin genomista edustaa lyhyitä DNA-toistoja, jotka koostuvat 20 emäsparista, ja suurin osa liikkuvista DNA-elementeistä (transposoneista) sijaitsee intergeenisillä alueilla. Tiedot riisin genomin koosta vaihtelevat kuitenkin merkittävästi.
Japanilaisella alalajilla genomin kooksi on määritetty 466 miljoonaa emäsparia ja Intian alalajilla 420 miljoonaa. Syy tähän eroon ei ole selvä. Se voi olla seurausta erilaisista metodologisista lähestymistavoista genomien ei-koodaavan osan koon määrittämisessä, eli se ei heijasta asioiden todellista tilaa. Mutta on mahdollista, että tutkittujen genomien koossa on 15 prosentin ero.
Toinen suuri ero paljastui löydettyjen geenien määrässä: japanilaisessa alalajissa 46 022 - 55 615 geeniä genomia kohden ja intialaisessa alalajissa 32 000 - 50 000. Syy tähän eroon ei ole selvä.
Saatujen tietojen epätäydellisyys ja epäjohdonmukaisuus mainitaan julkaistujen artikkeleiden kommenteissa. Tässä ilmaistaan myös toive, että puutteet riisin genomin tietämyksessä poistetaan vertaamalla "karkean sekvensoinnin" tietoja kansainvälisen riisin genomiprojektin osallistujien suorittaman yksityiskohtaisen, hierarkkisen sekvensoinnin tuloksiin.
VERTAILEVA JA TOIMINNALLINEN KASVIGENOMIIKKA
Saatu laaja tieto, josta puolet (kiinalaisen ryhmän tulokset) on julkisesti saatavilla, avaa epäilemättä laajat näkymät sekä riisin genomin tutkimukselle että kasvigenomiikan tutkimukselle yleensä. Arabidopsiksen ja riisin genomien ominaisuuksien vertailu osoitti, että suurin osa Arabidopsis-genomissa tunnistetuista geeneistä (jopa 80 %) löytyy myös riisin genomista, mutta noin puolet riisistä löytyvistä geeneistä vastaavat ( ortologeja) ei ole vielä löydetty Arabidopsiksen genomista. Samaan aikaan 98 % geeneistä, joiden primäärirakenne on todettu muille viljoille, löytyi riisin genomista.
Merkittävä (melkein kaksinkertainen) ero riisin ja Arabidopsiksen geenien lukumäärän välillä on hämmentävää. Samanaikaisesti kokonaissekvensoinnilla saatuja riisin genomin dekoodauksen luonnostietoja ei käytännössä verrata riisin genomin tutkimuksen laajoihin tuloksiin hierarkkisen kloonauksen ja sekvensoinnin menetelmällä, eli siihen, mikä on ei ole tehty Drosophilan genomin suhteen. Siksi on epäselvää, heijastaako Arabidopsiksen ja riisin geenien lukumäärän ero asioiden todellista tilaa vai johtuuko se metodologisten lähestymistapojen eroista.
Toisin kuin Arabidopsiksen genomissa, riisin genomin kaksoisgeeneistä ei anneta tietoja. On mahdollista, että niiden suhteellinen määrä voi olla suurempi riisissä kuin Arabidopsisissa. Tätä mahdollisuutta tukevat epäsuorasti tiedot riisin polyploidisten muotojen esiintymisestä. Lisää selvyyttä tähän asiaan voidaan odottaa, kun kansainvälinen riisigenomiprojekti on saatu päätökseen ja yksityiskohtainen kuva tämän genomin primääri-DNA-rakenteesta on saatu. Vakavia perusteita tällaiselle toivolle antaa se, että riisin genomin karkeaa sekvensointia koskevien töiden julkaisemisen jälkeen tämän genomin rakennetta koskevien julkaisujen määrä on lisääntynyt jyrkästi, erityisesti on ilmestynyt tietoa yksityiskohtaisesta sekvensoinnista. sen 1 ja 4 kromosomista.
Kasvien geenien lukumäärän ainakin likimääräinen tietäminen on perustavanlaatuista vertailevan kasvigenomiikan kannalta. Aluksi uskottiin, että koska kaikki kukkivat kasvit ovat fenotyyppisiltä ominaisuuksiltaan hyvin lähellä toisiaan, myös niiden genomien tulisi olla samanlaisia. Ja jos tutkimme Arabidopsiksen genomia, saamme tietoa useimmista muiden kasvien genomeista. Epäsuora vahvistus tälle olettamukselle on hiiren genomin sekvensoinnin tulokset, joka on yllättävän lähellä ihmisen genomia (noin 30 tuhatta geeniä, joista vain 1 tuhat osoittautui erilaiseksi).
Voidaan olettaa, että syy Arabidopsiksen ja riisin genomien välisiin eroihin piilee niiden kuulumisessa eri kasviluokkiin - kaksisirkkaisiin ja yksisirkkaisiin. Tämän asian selventämiseksi on erittäin toivottavaa tietää ainakin jonkin muun yksisirkkaisen kasvin karkea primaarirakenne. Todellisin ehdokas voisi olla maissi, jonka genomi on suunnilleen sama kuin ihmisen genomi, mutta silti paljon pienempi kuin muiden viljojen genomit. Maissin ravintoarvo tunnetaan hyvin.
Arabidopsiksen ja riisin genomien sekvensoinnin tuloksena saadusta valtavasta materiaalista on vähitellen tulossa perusta laajamittaiselle kasvien genomien tutkimukselle vertailevaa genomiikkaa käyttäen. Tällaisilla tutkimuksilla on yleinen biologinen merkitys, koska niiden avulla voidaan selvittää kasvin genomin kokonaisuuden ja yksittäisten kromosomien järjestäytymisen pääperiaatteet, tunnistaa geenien ja niiden säätelyalueiden rakenteen yhteiset piirteet ja ottaa huomioon kromosomin toiminnallisesti aktiivisen (geeni)osan ja erilaisten intergeenisten DNA-alueiden suhde, jotka eivät koodaa proteiineja. Vertailevasta genetiikasta on tulossa yhä tärkeämpää myös ihmisen toiminnallisen genomiikan kehittämisessä. Pufferfish- ja hiiren genomien sekvensointi suoritettiin vertailevia tutkimuksia varten.
Yhtä tärkeää on tutkia yksittäisiä geenejä, jotka vastaavat yksittäisten proteiinien synteesistä, jotka määrittävät kehon tietyt toiminnot. Ihmisgenomiohjelman käytännön, ensisijaisesti lääketieteellinen merkitys piilee yksittäisten geenien löytämisessä, eristämisessä, sekvensoinnissa ja toiminnan määrittämisessä. Tämän seikan pani merkille useita vuosia sitten J. Watson, joka korosti, että Human Genome -ohjelma saataisiin päätökseen vasta, kun kaikkien ihmisen geenien toiminnot selvitetään.

Riisi. 4. Luokittelu Arabidopsis-geenien toiminnan mukaan
1 - geenit kasvua, jakautumista ja DNA-synteesiä varten; 2 - RNA-synteesigeenit (transkriptio); 3 - geenit proteiinien synteesiä ja modifiointia varten; 4 - geenit kehitystä, ikääntymistä ja solukuolemaa varten; 5 - solujen aineenvaihdunnan ja energia-aineenvaihdunnan geenit; 6 - solujen välisen vuorovaikutuksen ja signaalinsiirron geenit; 7 - geenit muiden soluprosessien aikaansaamiseksi; 8 - geenit, joilla on tuntematon toimintaMitä tulee kasvigeenien toimintaan, tiedämme alle kymmenesosan siitä, mitä tiedämme ihmisen geeneistä. Jopa Arabidopsiksessa, jonka genomia on tutkittu paljon enemmän kuin ihmisen genomia, lähes puolet sen geeneistä on tuntematon (kuva 4). Samaan aikaan kasveilla on eläimille yhteisten geenien lisäksi huomattava määrä vain (tai ainakin pääosin) niille spesifisiä geenejä. Puhumme geeneistä, jotka osallistuvat veden kuljetukseen ja eläimillä puuttuvan soluseinän synteesiin, geeneistä, jotka varmistavat kloroplastien muodostumisen ja toiminnan, fotosynteesin, typen sitoutumisen sekä lukuisten aromaattisten tuotteiden synteesin. Tätä listaa voidaan jatkaa, mutta jo nyt on selvää, kuinka vaikea tehtävä kasvien toiminnallisella genomiikalla on edessään.
Koko genomin sekvensointi antaa lähes todellista tietoa tietyn organismin geenien kokonaismäärästä, mahdollistaa enemmän tai vähemmän yksityiskohtaisen ja luotettavan tiedon sijoittamisen niiden rakenteesta tietopankkeihin sekä helpottaa yksittäisten geenien eristämistä ja tutkimista. Genomisekvensointi ei kuitenkaan suinkaan tarkoita kaikkien geenien toiminnan vahvistamista.
Yksi toiminnallisen genomiikan lupaavimmista lähestymistavoista perustuu toimivien geenien tunnistamiseen, joita käytetään mRNA:n transkriptioon (lukemiseen). Tämä lähestymistapa, mukaan lukien nykyaikaisen mikrosiruteknologian käyttö, mahdollistaa jopa kymmenien tuhansien toimivien geenien tunnistamisen samanaikaisesti. Äskettäin tätä lähestymistapaa käyttäen on aloitettu kasvien genomien tutkimus. Arabidopsikselle oli mahdollista saada noin 26 tuhatta yksittäistä transkriptiota, mikä helpottaa suuresti mahdollisuutta määrittää melkein kaikkien sen geenien toiminta. Perunoista pystyttiin tunnistamaan noin 20 000 toimivaa geeniä, jotka ovat tärkeitä sekä kasvu- ja mukuloiden muodostumisprosessien että perunatautiprosessien ymmärtämiselle. Tämän tiedon oletetaan lisäävän yhden tärkeimmistä elintarvikkeista vastustuskykyä taudinaiheuttajia vastaan.
Funktionaalisen genomiikan looginen kehitys oli proteomiikkaa. Tämä uusi tieteenala tutkii proteomia, joka yleensä ymmärretään täydellisenä proteiinisarjana solussa tietyllä hetkellä. Tällainen genomin toiminnallista tilaa heijastava proteiinisarja muuttuu koko ajan, kun taas genomi pysyy muuttumattomana.
Proteiinien tutkimusta on käytetty pitkään arvioimaan kasvien genomien aktiivisuutta. Kuten tiedetään, kaikissa kasveissa esiintyvät entsyymit eroavat toisistaan yksittäisten lajien ja lajikkeiden osalta aminohappojen sekvenssissä. Tällaisia entsyymejä, joilla on sama toiminto, mutta erilainen yksittäisten aminohappojen sekvenssi, kutsutaan isoentsyymeiksi. Niillä on erilaisia fysikaalis-kemiallisia ja immunologisia ominaisuuksia (molekyylipaino, varaus), jotka voidaan havaita kromatografialla tai elektroforeesilla. Näitä menetelmiä on useiden vuosien ajan käytetty menestyksekkäästi tutkittaessa ns. geneettistä polymorfismia eli organismien, lajikkeiden, populaatioiden, lajien, erityisesti vehnän ja siihen liittyvien viljamuotojen välisiä eroja. Viime aikoina DNA-analyysimenetelmien, mukaan lukien sekvensoinnin, nopean kehityksen vuoksi proteiinipolymorfismin tutkimus on kuitenkin korvattu DNA-polymorfismin tutkimuksella. Viljan tärkeimmät ravitsemukselliset ominaisuudet määrittävien varastoproteiinien (prolamiinit, gliadiinit jne.) spektrien suora tutkimus on kuitenkin edelleen tärkeä ja luotettava menetelmä maatalouskasvien geneettiseen analyysiin, valintaan ja siementuotantoon.
Geenien, niiden ilmentymis- ja säätelymekanismien tuntemus on erittäin tärkeää biotekniikan kehittämisen ja siirtogeenisten kasvien tuotannon kannalta. Tiedetään, että vaikuttavat menestykset tällä alalla aiheuttavat moniselitteisen reaktion ympäristö- ja lääketieteellisessä yhteisössä. Kasvibiotekniikassa on kuitenkin alue, jolla nämä pelot, elleivät täysin perusteettomia, näyttävät joka tapauksessa olevan vähäisiä. Puhumme siirtogeenisten teollisuuslaitosten luomisesta, joita ei käytetä elintarviketuotteina. Intiassa korjattiin äskettäin ensimmäinen sato siirtogeenistä puuvillaa, joka on vastustuskykyinen useille taudeille. Tietoa on erityisten pigmenttiproteiineja koodaavien geenien tuomisesta puuvillan genomiin ja sellaisten puuvillakuitujen tuotannosta, jotka eivät vaadi keinotekoista värjäystä. Toinen teollinen viljelykasvi, joka voi olla tehokkaan geenitekniikan kohteena, on pellava. Sen käytöstä vaihtoehtona puuvillalle tekstiilien raaka-aineissa on keskusteltu viime aikoina. Tämä ongelma on erittäin tärkeä maallemme, joka on menettänyt omat raakapuuvillan lähteensä.
NÄKYMÄT KASVIGENOMIEN TUTKIMUKSESTA
On selvää, että kasvien genomien rakennetutkimukset perustuvat vertailevan genomiikan lähestymistapoihin ja menetelmiin, joissa päämateriaalina käytetään Arabidopsiksen ja riisin genomien purkamisen tuloksia. Tärkeä rooli vertailevan kasvigenomiikan kehittämisessä tulee epäilemättä olemaan tiedolla, joka ennemmin tai myöhemmin saadaan muiden kasvien genomien totaalisella (karkealla) sekvensoinnilla. Tässä tapauksessa vertaileva kasvigenomiikka perustuu geneettisten suhteiden selvittämiseen yksittäisten lokusten ja eri genomeihin kuuluvien kromosomien välillä. Emme keskity niinkään kasvien yleiseen genomiikkaan kuin yksittäisten kromosomaalisten lokusten selektiiviseen genomiikkaan. Esimerkiksi hiljattain on osoitettu, että vernaalisaatiosta vastaava geeni sijaitsee heksaploidisen vehnäkromosomin 5A VRn-AI-lokuksessa ja riisin kromosomin 3 Hd-6-lokuksessa.
Näiden tutkimusten kehittäminen on voimakas sysäys monien toiminnallisesti tärkeiden kasvigeenien tunnistamiseen, eristämiseen ja sekvensointiin, erityisesti geenien, jotka vastaavat taudinkestävyydestä, kuivuuden kestävyydestä ja sopeutumiskyvystä erilaisiin kasvuolosuhteisiin. Yhä enemmän käytetään toiminnallista genomiikkaa, joka perustuu kasveissa toimivien geenien massadetektioon (seulomiseen).
Voimme ennakoida kromosomitekniikoiden, ensisijaisesti mikrodissektiomenetelmän, parantamista edelleen. Sen käyttö laajentaa dramaattisesti genomitutkimuksen mahdollisuuksia ilman suuria kustannuksia, kuten esimerkiksi genomin kokonaissekvensointia. Menetelmä lokalisoida yksittäisten geenien kasvien kromosomeihin hybridisaation avulla leviää edelleen. paikan päällä. Tällä hetkellä sen käyttöä rajoittaa valtava määrä toistuvia sekvenssejä kasvin genomissa ja mahdollisesti kasvien kromosomien rakenteellisen järjestyksen erityispiirteet.
Kromosomitekniikoista tulee lähitulevaisuudessa suuri merkitys kasvien evoluutiogenomiikan kannalta. Nämä suhteellisen edulliset tekniikat mahdollistavat nopean sisäisen ja interspesifisen vaihtelun arvioinnin, tetraploidisen ja heksaploidisen vehnän, ruisvehnän kompleksisten allopolyploidisten genomien tutkimisen; analysoida evoluutioprosesseja kromosomitasolla; tutkia synteettisten genomien muodostumista ja vieraan geneettisen materiaalin sisääntuloa (introgressiota); tunnistaa geneettisiä suhteita eri lajien yksittäisten kromosomien välillä.
Genomin karakterisoinnissa käytetään kasvien karyotyypin tutkimusta klassisilla sytogeneettisillä menetelmillä, molekyylibiologisella analyysillä ja tietokonetekniikalla rikastettuna. Tämä on erityisen tärkeää tutkittaessa karyotyypin stabiilisuutta ja vaihtelua ei vain yksittäisten organismien, vaan myös populaatioiden, lajikkeiden ja lajien tasolla. Lopuksi on vaikea kuvitella, kuinka kromosomien uudelleenjärjestelyjen (poikkeamat, sillat) lukumäärä ja spektrit voidaan arvioida ilman differentiaalivärjäysmenetelmiä. Tällaiset tutkimukset ovat erittäin lupaavia ympäristön seurannassa kasvin genomin tilan perusteella.
Nyky-Venäjällä kasvien genomien suoraa sekvensointia ei todennäköisesti tehdä. Tällainen suuria investointeja vaativa työ on nykyisen taloutemme vahvuuksien ulkopuolella. Samaan aikaan maailmantieteen hankkimat ja kansainvälisistä tietopankeista saatavilla olevat tiedot Arabidopsiksen ja riisin genomien rakenteesta riittävät kotimaisen kasvigenomiikan kehittämiseen. Voidaan ennakoida vertaileviin genomiikan lähestymistapoihin perustuvien kasvigenomitutkimusten laajentamista jalostuksen ja kasvinviljelyn erityisongelmien ratkaisemiseksi sekä erilaisten taloudellisesti merkittävien kasvilajien alkuperän tutkimiseksi.
Voidaan olettaa, että meidän budjetillemme varsin edullisia genomisia lähestymistapoja, kuten geneettistä tyypitystä (RELF, RAPD, AFLP-analyysit jne.), käytetään laajasti kotimaisessa jalostuksessa ja kasvinviljelyssä. DNA-polymorfismin suorien määritysmenetelmien rinnalla genetiikan ja kasvinjalostuksen ongelmien ratkaisemisessa käytetään proteiinipolymorfismin, ensisijaisesti viljan varastoproteiinien, tutkimukseen perustuvia lähestymistapoja. Kromosomiteknologiaa tullaan käyttämään laajalti. Ne ovat suhteellisen edullisia, niiden kehittäminen vaatii melko maltillisia investointeja. Kromosomitutkimusten alalla kotimainen tiede ei ole huonompi kuin maailma.
On korostettava, että tieteemme on antanut merkittävän panoksen kasvien genomiikan muodostumiseen ja kehitykseen [ , ].
Pääroolin näytteli N.I. Vavilov (1887-1943).
Molekyylibiologiassa ja kasvigenomiikassa uraauurtava panos A.N. Belozersky (1905-1972).
Kromosomitutkimusten alalla on huomioitava erinomaisen geneetiikan S.G. Navashin (1857-1930), joka löysi ensimmäisenä satelliittikromosomit kasveista ja osoitti, että on mahdollista erottaa yksittäiset kromosomit niiden morfologian ominaisuuksien mukaan.
Toinen venäläisen tieteen klassikko G.A. Levitsky (1878-1942) kuvasi yksityiskohtaisesti rukiin, vehnän, ohran, herneiden ja sokerijuurikkaan kromosomit, otti käyttöön termin "karyotyyppi" tieteeseen ja kehitti sen opin.
Nykyaikaiset asiantuntijat, jotka luottavat maailmantieteen saavutuksiin, voivat antaa merkittävän panoksen kasvigenetiikan ja genomiikan jatkokehitykseen.
Kirjoittaja ilmaisee sydämelliset kiitoksensa akateemikolle Yu.P. Altukhov kriittisestä keskustelusta artikkelista ja arvokkaista neuvoista.Artikkelin kirjoittajan johtaman ryhmän työtä tuki Venäjän perustutkimussäätiö (apurahat nro 99-04-48832; 00-04-49036; 00-04-81086), presidentin ohjelma. Venäjän federaatio tieteellisten koulujen tukemiseksi (apurahat 00-115 -97833 ja NSh-1794.2003.4) ja Venäjän tiedeakatemian ohjelma "Molekulaariset geneettiset ja kromosomaaliset markkerit nykyaikaisten jalostus- ja siemententuotantomenetelmien kehittämisessä" .
KIRJALLISUUS
1. Zelenin A.V., Badaeva E.D., Muravenko O.V. Johdatus kasvien genomiikkaan // Molekyylibiologia. 2001. V. 35. S. 339-348. 2. Kynä E. Bonanza kasvien genomiikalle // Tiede. 1998. V. 282. P. 652-654. 3. Plant genomics, Proc. Natl. Acad. sci. USA. 1998. V. 95. P. 1962-2032. 4. Kartelli N.A. jne. Genetiikka. Ensyklopedinen sanakirja. Minsk: Technologia, 1999. 5. Badaeva E.D., Friebe B., Gill B.S. 1996. Genomin erilaistuminen Aegilopsissa. 1. Erittäin toistuvien DNA-sekvenssien jakautuminen diploidisten lajien kromosomeissa, genomi. 1996. V. 39. S. 293-306. Kromosomianalyysin historia // Biol. kalvot. 2001. T. 18. S. 164-172.




